

«El todo es más que la suma de las partes»
Aristóteles«La gloria de la medicina es que avanza constantemente, que siempre hay algo más por aprender»
W.J. MayoLa miocardiopatía hipertrófica (MCH) es una causa importante de discapacidad y muerte de pacientes de todas las edades, pero la muerte súbita inesperada de adultos jóvenes es el componente más espantoso de su evolución natural. El trastorno se produce principalmente por mutaciones en genes sarcoméricos, pero hay también otros factores que pueden modular el fenotipo; de hecho, muchos de los portadores de mutaciones patogénicas/causales no muestran la enfermedad.
El objetivo del elegante estudio de Pérez-Sánchez et al.1 recientemente publicado en Revista Española de Cardiología fue determinar si factores como el sexo, la hipertensión y la actividad física modifican la gravedad de la enfermedad, así como establecer su papel en la penetrancia según edad de la MCH o la prevalencia de casos de la enfermedad en portadores de mutaciones causales, en una cohorte amplia de familias con MCH genotipificadas que presentaban mutaciones en los genes sarcoméricos MYBPC3 y MHY7, que son las mutaciones causales más frecuentes. El estudio de esta cohorte permitió detectar diferencias en el fenotipo pero evitó incluir a pacientes con mutaciones múltiples que causan fenotipos más desfavorables. Es probable que la reducción de la penetrancia se deba a combinaciones de diversos factores genéticos y ambientales externos e internos (bernardianos)2.
La MCH es la cardiopatía genética supuestamente monogénica más difundida, con una prevalencia global del 0,2% (≈ 700.000 casos en Estados Unidos), pero no se conoce la prevalencia de las mutaciones patogénicas específicas. El descubrimiento de mutaciones sarcoméricas como base genética de la MCH ha abierto nuevas vías importantes para el conocimiento de su patogenia a escala molecular, incluidas la contribución de la disfunción mitocondrial y las anomalías energéticas que conlleva.
Desde el punto de vista histopatológico, la MCH es una enfermedad miocárdica que se manifiesta por una matriz intersticial expandida, un agrandamiento de los núcleos de los miocitos, un engrosamiento y desalineación/desorganización de las miofibras, así como una hipertrofia, generalmente asimétrica, de la pared del ventrículo izquierdo (HVI), incluidos los músculos papilares y las trabéculas. La HVI es una secuela directa de las mutaciones de la MCH y no es compensatoria/secundaria a una sobrecarga de presión3. Se asocia con una limitación del tamaño de la cavidad del ventrículo izquierdo (VI), pero en las fases terminales de la enfermedad aparecen insuficiencia cardiaca y dilatación ventriculoauricular, con adelgazamiento de la pared y cicatrización.
CATETERIZACIÓN CON MULTISENSORES-FENOTIPO DE LA MCHMediante cateterización micromanométrica/velocimétrica con multisensores, junto con las modalidades de diagnóstico por imagen digital cardiaca, actualmente resulta posible establecer nuevas correlaciones hemodinámicas cuantitativas entre genotipo y fenotipo (estudios de asociación de genoma completo [GWAS]) de la MCH. Es necesaria una relación predecible entre el genotipo y la expresión de la enfermedad de MCH para que se pueda aplicar pruebas genéticas útiles para la toma de decisiones clínicas4. Los estudios multimodales brindan actualmente la perspectiva de identificar cambios en múltiples facetas del fenotipo de la MCH en cuanto a la dinámica sistólica y diastólica3, en un momento en que el avance constante del conocimiento resulta más imperativo que en la época de Mayo (epigrama).
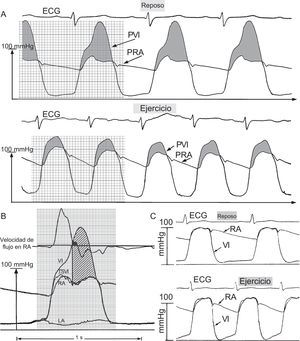
El deterioro de la dinámica diastólica y la hiperdinamia sistólica son características distintivas espectaculares de la caracterización de la MCH, sobre todo durante el ejercicio, tal como se resume en la figura 1. La MCH manifiesta lo que en publicaciones anteriores he denominado «gradientes de presión polimórficos»3,5,6. A pesar de que las guías clínicas recomiendan evitar el ejercicio intenso/de competición, la asociación entre la actividad física y los resultados clínicos de la MCH parece débil1.
 profundo y de la raíz aórtica (PRA) en la miocardiopatía hipertrófica en reposo y durante el ejercicio de bicicleta en decúbito supino, que provoca una disminución diastólica anormal de la PVI, lo cual indica deterioro de la relajación ventricular; la disminución de la PVI durante toda la diástole contrasta claramente con el patrón normal que se muestra en el panel C. B: relación presión-flujo con altos gradientes dinámicos mesosistólicos y telesistólicos tempranos y enormes en la miocardiopatía hipertrófica; de arriba abajo, señal de velocidad aórtica y señales micromanométricas del ventrículo izquierdo (VI) profundo, el tracto de salida del VI (TSVI) y la raíz aórtica (RA), medidas con un catéter cardiaco izquierdo con multisensores de presión y velocidad retrógrada de triple punta; la señal micromanométrica de la aurícula izquierda (AI) se determinó al mismo tiempo mediante un catéter transeptal; la línea recta vertical identifica el inicio del contacto entre el movimiento sistólico anterior y el tabique, determinado mediante una ecocardiografía de la válvula mitral en modo M simultánea (no mostrada); la mayor parte del flujo de eyección aórtico se ha completado ya en ese momento; el enorme gradiente mesosistólico y telesistólico (área sombreada) se mantienen ante la minúscula velocidad aórtica anterógrada restante o incluso negativa. Adaptado de Pasipoularides3 con permiso de PMPH-USA.")
A: presión del ventrículo izquierdo (PVI) profundo y de la raíz aórtica (PRA) en la miocardiopatía hipertrófica en reposo y durante el ejercicio de bicicleta en decúbito supino, que provoca una disminución diastólica anormal de la PVI, lo cual indica deterioro de la relajación ventricular; la disminución de la PVI durante toda la diástole contrasta claramente con el patrón normal que se muestra en el panel C. B: relación presión-flujo con altos gradientes dinámicos mesosistólicos y telesistólicos tempranos y enormes en la miocardiopatía hipertrófica; de arriba abajo, señal de velocidad aórtica y señales micromanométricas del ventrículo izquierdo (VI) profundo, el tracto de salida del VI (TSVI) y la raíz aórtica (RA), medidas con un catéter cardiaco izquierdo con multisensores de presión y velocidad retrógrada de triple punta; la señal micromanométrica de la aurícula izquierda (AI) se determinó al mismo tiempo mediante un catéter transeptal; la línea recta vertical identifica el inicio del contacto entre el movimiento sistólico anterior y el tabique, determinado mediante una ecocardiografía de la válvula mitral en modo M simultánea (no mostrada); la mayor parte del flujo de eyección aórtico se ha completado ya en ese momento; el enorme gradiente mesosistólico y telesistólico (área sombreada) se mantienen ante la minúscula velocidad aórtica anterógrada restante o incluso negativa. Adaptado de Pasipoularides3 con permiso de PMPH-USA.
He elaborado un modelo del flujo que permite esclarecer la hemodinámica sistólica tardía de la MCH con eliminación en la cavidad3,5,6. Continúa habiendo controversia respecto a si el contacto de la valva mitral con el tabique causa obstrucción al flujo de salida y el enorme gradiente intraventricular mesosistólico y telesistólico (figura 1B). Lo describió Braunwald7, el principal pionero en el campo de la MCH, bajo cuyo inspirado liderazgo tuve el privilegio de trabajar, al inicio de mi carrera, en el Departamento de Medicina del Peter Bent Brigham Hospital, en Harvard.
Las anomalías coexistentes con una función diastólica biventricular prominente se muestran también en la figura 1, sobre todo durante el ejercicio físico agudo3,8. Serán necesarios más estudios sobre la interacción de los factores3 que subyacen a la dinámica del flujo de entrada del VI9 y la pronunciada heterogeneidad existente en las contribuciones relativas de los defectos de relajación10, la asincronía11 y la alteración de las propiedades diastólicas pasivas y la geometría12,13 a la disfunción diastólica general presente en la MCH.
CONTEXTO GENÉTICO/GENÓMICO DE LA MCHLa MCH tiene variantes genéticas causantes de la enfermedad y muestra heterogeneidad fenotípica interfamiliar e intrafamiliar en los aspectos morfológicos, funcionales, clínicos y pronósticos. Sin embargo, las inconcordancias intrafamiliares no se explican por la heterogeneidad de las mutaciones; por consiguiente, también deben de estar involucrados factores ambientales. La penetrancia reducida/incompleta y la expresividad variable (plasticidad fenotípica) de la mutación o mutaciones patogénicas/causales han impedido alcanzar un conocimiento integral de su espectro clínico. La diversidad fenotípica entre los individuos se debe a diferencias en la secuencia del ADN y a influencias ambientales internas/externas en genes que tienen una expresividad variable/penetrancia incompleta3. Una combinación de factores genéticos, ambientales, aleatorios y de estilo de vida hace que la probabilidad de que el fenotipo de la enfermedad se manifieste en los portadores de genes de la MCH sea < 1.
La secuenciación del genoma humano ha permitido establecer el claro vínculo entre un número creciente de enfermedades, incluida la MCH, y numerosas variantes polimórficas genéticas/genómicas, tal como se resume en el icónico catálogo GWAS del United States National Human Genome Research Institute y el European Bioinformatics Institute (NHGRI-EBI) en la figura 2. El diagrama se actualiza cada noche y puede consultarse la versión más reciente en la página web del NHGRI-EBI Catalog14. La mayoría de las mutaciones de la MCH son «privadas», es decir, específicas de una familia. La MCH es causada por mutaciones en genes que contienen el código principalmente de proteínas del aparato contráctil sarcomérico15; la mayoría de ellas son mutaciones antisentido que consisten en la sustitución de un aminoácido por otro. Estas mutaciones modifican propiedades físicas/funcionales de las proteínas incorporadas a los sarcómeros y pueden inducir señales hipertróficas. Con menor frecuencia, aparecen inserciones o deleciones de nucleótidos en las mutaciones de desplazamiento de marco, que alteran profundamente la traducción al ARN mensajero y las propiedades de las proteínas resultantes.
 y el European Bioinformatics Institute (EMBL-EBI) han proporcionado conjuntamente el Genome-wide Association Studies (GWAS) Catalog. El diagrama se actualiza cada noche y se puede consultar la versión más reciente en la página web del NHGRI-EBI Catalog14. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
Diagrama que muestra todas las asociaciones de polimorfismos de un solo nucleótido con rasgos que tienen un valor de p ≤ 5,0 × 10–8 en un mapa del genoma humano con las localizaciones cromosómicas y presentado en el cariotipo humano. El National Human Genome Research Institute (NHGRI) y el European Bioinformatics Institute (EMBL-EBI) han proporcionado conjuntamente el Genome-wide Association Studies (GWAS) Catalog. El diagrama se actualiza cada noche y se puede consultar la versión más reciente en la página web del NHGRI-EBI Catalog14. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
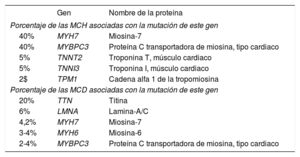
Mediante inferencia a partir de las listas de pruebas de paneles multigénicos de MCH actualmente comercializados16, hasta ahora se han identificado mutaciones patógenas/causales en ≈25–30 genes, entre los que se encuentran los siguientes: MYBPC3, MYH7, TNNI3, TNNT2, MTP, MYL2, MYL3, ACTC, ACTN2 y TCAP. Es de destacar que las mutaciones de los genes MYH7 y MYBPC3 suponen ≈ 80% de los casos genotipificados, tal como se muestra en la tabla15,17.
Genes involucrados en diversas miocardiopatías, en orden decreciente de frecuencia
| Gen | Nombre de la proteína | |
|---|---|---|
| Porcentaje de las MCH asociadas con la mutación de este gen | ||
| 40% | MYH7 | Miosina-7 |
| 40% | MYBPC3 | Proteína C transportadora de miosina, tipo cardiaco |
| 5% | TNNT2 | Troponina T, músculo cardiaco |
| 5% | TNNI3 | Troponina I, músculo cardiaco |
| 2$ | TPM1 | Cadena alfa 1 de la tropomiosina |
| Porcentaje de las MCD asociadas con la mutación de este gen | ||
| 20% | TTN | Titina |
| 6% | LMNA | Lamina-A/C |
| 4,2% | MYH7 | Miosina-7 |
| 3-4% | MYH6 | Miosina-6 |
| 2-4% | MYBPC3 | Proteína C transportadora de miosina, tipo cardiaco |
Estos genes contienen el código para la síntesis de proteínas sarcoméricas, el disco Z y la regulación del calcio (la señalización del Ca2+ es un desencadenante esencial de la hipertrofia) con una patogenicidad diversa. Todos ellos intensifican la sensibilidad/señalización del Ca2+ y la función de los miofilamentos, lo que da lugar a una hipercontractilidad miocitaria que aumenta la generación de fuerza sarcomérica y la velocidad de deslizamiento de la actina-miosina. Genera un aumento de la hidrolisis de la adenosina trifosfato (ATP) y un balance de energía negativo; con el tiempo, esto conduce a una disfunción diastólica3.
La existencia de varios síndromes hereditarios con deterioro de la producción de energía mitocondrial e hipertrofia cardiaca asimétrica que son clínicamente indistinguibles de la MCH indica que el déficit de energía es esencial para producir el fenotipo de la MCH. Las variantes génicas/alelos que producen estas fenocopias de la MCH (es decir, rasgos típicos de otro genotipo) son las de la enfermedad de Fabry (GLA), la enfermedad de Danon (LAMP2), la enfermedad de Pompe (GAA), la enfermedad de almacenamiento de glucógeno (PRKAG2) y otras enfermedades ligadas a defectos de la cadena respiratoria mitocondrial (ya mencionadas). Las pruebas genéticas las diferencian de la MCH.
Las relaciones anormales entre Ca2+ y ATP subyacen a la fisiopatología/anatomopatología de la MCHComo ya se ha comentado, la sensibilización al Ca2+ de las mutaciones ligadas a la MCH eleva la contractilidad sistólica, pero conduce a una alteración de la regulación del Ca2+ que deteriora la relajación de los miocardiocitos, lo cual se debe a una reducción de la frecuencia de los ciclos de puentes cruzados y, por consiguiente, deriva directamente de las mutaciones de la MCH. La relajación/disfunción diastólica debe contribuir a la incidencia de muerte súbita en la MCH, a causa de arritmias ventriculares mortales o a concentraciones anormalmente altas del Ca2+ citosólico libre. Estas elevaciones del Ca2+ tienen efectos nocivos, en parte por la sobrecarga de las mitocondrias con Ca2+ que causa una disminución de la formación de ATP como consecuencia de la despolarización de la membrana interna mitocondrial18. La notable reducción de la generación de ATP en las mitocondrias impide que las bombas de Ca2+, que se encuentran en el sarcolema y la red del retículo sarcoplásmico alrededor de cada miofibrilla, eliminen el exceso de Ca2+ del citosol, con lo que se crea un círculo vicioso. Probablemente, algún aumento del estímulo simpático cardiaco inducido por el ejercicio o el estrés debe de acentuarlo.
Además, la elevación del Ca2+ citosólico libre, en especial durante la diástole, intensificaría la hipertrofia miocardiocitaria a través de la activación de diversas vías de señalización que modifican la expresión genética de manera dependiente del Ca2+, incluida la calcineurina/NFAT/GATA4 y la cinasa II dependiente de calmodulina19–21. La consiguiente depleción relativa/absoluta de ATP agrava el deterioro de la recaptación de Ca2+ en el retículo sarcoplásmico y la disfunción diastólica3,12.
LA MCH EN LA ERA DE LA GENÓMICA: VARIABILIDAD GENOTÍPICA Y FENOTÍPICALa genética y la biología que subyacen a la MCH se encuentran actualmente en un punto de inflexión de gran interés. Las rápidas innovaciones en las tecnologías de determinación del genotipo aparecidas en los últimos años han permitido la realización de GWAS para la identificación de nuevos genes candidatos para la MCH correlacionados con la susceptibilidad a la enfermedad15.
Para descifrar las repercusiones funcionales/morfomecánicas de las variantes polimórficas presentes en el genoma humano, es necesario determinar sus efectos en la expresión génica. Sin embargo, la expresión de mutaciones activas probablemente fluctúe con el paso del tiempo. Los genes pueden estar regulados, es decir, activarse e inactivarse en los momentos oportunos y en contextos apropiados, por pistas/señales específicas procedentes de fuera del genoma y que interaccionan con «interruptores» del ADN promotores, operadores y silenciadores situados a lo largo de los genes, que los controlan. Esta regulación utiliza factores de transcripción, que son proteínas que se unen al ADN en determinadas secuencias diana y hacen que resulte más difícil o más fácil la unión de la ARN polimerasa al promotor del gen y el inicio de la transcripción. Actúan como reóstatos, de tal manera que, en función de la mayor o menor actividad del controlador, se obtiene más o menos producto/efecto. Este proceso generalmente permite a las células realizar operaciones lógicas y combinar diferentes fuentes de información en continuo cambio para «decidir» si expresan un gen o no; habitualmente permite a los genes adaptar su función a unas exigencias cambiantes y expresar de manera eficiente rasgos morfomecánicos distintos y modificables de un modo selectivo y dependiente de la actividad2,15.
Por consiguiente, la determinación del perfil de expresión génica en una sola ocasión en un individuo/paciente/familiar puede ser poco fiable para determinar el diagnóstico, el tratamiento o el pronóstico2. En la MCH son necesarias pruebas genéticas seriadas y, con los nuevos conocimientos, se avanza en el esclarecimiento de la causalidad y la patogenia de la enfermedad, dentro del rápido desarrollo de las bases de datos de bioinformática humana que mantienen las universidades/centros de investigación, como la base del Exome Aggregation Consortium (ExAC) y el International Genome Sample Resource (IGSR) 1000 Genomes. De hecho, los datos existentes son ya tantos que resulta imprescindible elaborar herramientas eficaces, con el empleo de inteligencia artificial con aprendizaje automático y redes neurales, que permitirán elaborar algoritmos de toma de decisión clínica capaces de seleccionar en las bases de datos la información útil.
¿Mecanismos de MCH poligénicos clasificados erróneamente como monogénicos? Una controversia cambianteEl resultado imperfecto de las pruebas genéticas en la MCH puede reflejar un conocimiento incompleto de todos los genes involucrados en la enfermedad, que se expresan dentro de un programa regulado. Una mutación crítica en el desarrollo inicial de la MCH puede dejar de ser activa luego, o mutaciones que están invariablemente activas en una fase pueden dejar de estarlo en fases posteriores de la evolución de la enfermedad2. Se producen, pues, reclasificaciones de mutaciones patogénicas «mal clasificadas» anteriormente, como consecuencia de la información genética y funcional de la que se dispone más tarde; con lo que también hay casos de familiares con genotipo positivo (G+) y fenotipo negativo (P–) que ilustrarían los efectos (poligénicos) de genes modificadores.
Así pues, tal como se ha propuesto recientemente15, parece probable que enfermedades como la MCH y mecanismos generadores de fenotipos poligénicos puedan clasificarse de manera inexacta como monogénicos. Ciertamente, hay precedentes en otras enfermedades relacionadas con la disfunción mitocondrial, como el síndrome de Barth que muestra una miocardiopatía dilatada o la enfermedad de Parkinson que inicialmente se consideró monogénica/mendeliana. Actualmente esto parece una simplificación excesiva, puesto que la diversidad fenotípica en los familiares implica la presencia de interacciones poligénicas y de factores ambientales/epigenéticos como importantes moduladores de la enfermedad. Estas acciones con múltiples facetas traen a la mente el holismo de Aristóteles, que se expresa en el epigrama «El todo es más que la suma de las partes». Los todos no son reducibles a sus partes ya que tienen propiedades emergentes, que proceden de nuevas interacciones entre los componentes. Los efectos ambientales en la expresión de la enfermedad pueden ser grandes, tal como pone de manifiesto la aparición de la enfermedad de Parkinson en algunos portadores de variantes expuestos a plaguicidas.
Existen centenares de mutaciones antisentido de MYH7 y MYBPC3 causantes de MCH, y las mismas variantes antisentido se encuentran también en la miocardiopatía dilatada (tabla), lo cual subraya la importancia del contexto genómico (genes modificadores que influyen en la expresión fenotípica y la gravedad de los alelos patogénicos/causales). Tanto la diversidad fenotípica interfamiliar como la intrafamiliar de cada mutación pueden explicarse, al menos en parte, por su base genética: hay una reformulación de la expresividad y la variabilidad fenotípica de las variantes causales que conduce a una pleotropía15. No obstante, las observaciones que indican variaciones intrafamiliares indican que, además de la heterogeneidad mutacional, deben intervenir también factores ambientales.
FALACIA REDUCCIONISTA, EPISTASIS Y CONCEPTO ARISTOTÉLICO DE HOLISMOAl explicar cosas complejas en términos de causas más sencillas, podemos caer en la falacia reduccionista: como A se vincula con B, A no es otra cosa que B; por ejemplo, una catedral no más que una pila de piedras; una sonata para violín no es más que una secuencia de cuerdas en vibración. Como apunta el holismo de Aristóteles, el todo es más que la suma de sus partes y contiene propiedades emergentes que no pueden deducirse a través de la exploración de las partes.
De manera análoga, la epistasis significa que el efecto o efectos de un gen dependen de la presencia de uno o varios genes modificadores (base/contexto genético); además, las mutaciones epistáticas combinadas tienen efectos diferentes que individualmente15. Así pues, las interacciones entre genes generan conjuntos más amplios de posibles genotipos, y sus correspondientes fenotipos. La epistasis es una causa importante de multifuncionalidad génica. Un aspecto importante de la epistasis es que no solo influye en el fenotipo, sino que oculta la presencia/efectos de otros genes. El gen de la calvicie total es epistático respecto a los del pelo rubio o rojo. Más allá de la epistasis, las interacciones entre gen y entorno multiplican aún más la diversidad fenotípica2,15,20,21.
Distinción poco clara entre herencia monogénica y herencia poligénica/complejaLas interacciones genéticas generan una variedad interindividual en los fenotipos (de enfermedad) y, además, hacen que sea poco clara la distinción entre la herencia monogénica y la digénica, poligénica o compleja, en las que es necesaria la interacción de mutaciones de 2 o más genes distintos para la expresión de fenotipos clínicos diversos2,15. Las enfermedades genéticas muy probablemente ilustren todo un espectro o progresión de influencia decreciente, que va de un único gen causal afectado por genes modificadores, hasta los múltiples genes covalentes con una potencia cada vez más parecida, pasando por todos los pasos intermedios. Parece claro que los genes modificadores o covalentes pueden modular ampliamente el fenotipo, lo que hace que el diagnóstico, el pronóstico y el tratamiento basados en el genotipo sean problemáticos.
Probablemente como consecuencia de diversas combinaciones de factores genéticos y ambientales internos/externos (incluido el estilo de vida), que en su mayoría no están identificados, la reducida penetrancia y la expresividad variable son fenómenos que afectan de manera característica a trastornos como la MCH, que históricamente se han clasificado (mal) como autosómicos dominantes. Hasta el momento, no se ha podido establecer un postulado poligénico/multifactorial correspondiente a la MCH; sin embargo, se acepta que, dependiendo de la mutación o las mutaciones concretas de que se trate, la penetrancia está en función de la edad, el sexo y la forma física/ejercicio; todos ellos son factores importantes que afectan también a la expresión fenotípica en la hipertrofia concéntrica secundaria3,20,21.
FACTORES NO GENÉTICOS QUE AFECTAN A LA EXPRESIÓN DE UNA MUTACIÓN DE MCHLa cardiología personalizada de hoy incluye la integración en la práctica clínica de estudios de alta tecnología genética/genómica y otros estudios de biología molecular2,3,15. La diversidad fenotípica que caracteriza la MCH indica una naturaleza multifactorial, y en la expresión de la enfermedad intervienen genes modificadores y factores ambientales internos/externos (figura 3). Sin embargo, Pérez-Sánchez et al.1 concluyen que, una vez aparecido el fenotipo de MCH, el sexo, la hipertensión y, muy especialmente, la actividad física no se asocian con la gravedad de la enfermedad ni tienen una repercusión importante en el pronóstico de los portadores de mutaciones causales.
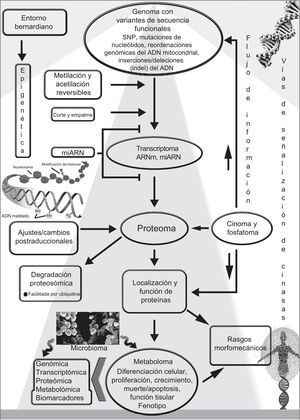
 y las vías de señalización en la transición y expresión de los genes humanos, mientras que la cantidad de proteínas y sus funciones se ven afectadas por diversos ajustes postraduccionales. Dado que los estados epigenéticos son reversibles, pueden modificarse por factores ambientales que pueden contribuir a cambios modificables entre los fenotipos normales y anormales. s posible que la interferencia en la actividad de factores que modifican el estado de la cromatina afecte a la expresión de variantes génicas no deseadas. El organismo humano es de hecho un «supraorganismo», es decir, una mezcla de genes y rasgos humanos y del microbioma, incluido el metaboloma. La relación entre los diferentes componentes «-oma/-ómicos», la cantidad de proteínas, la modificación postraduccional y la localización y la actividad de las proteínas que afectan a los rasgos fenotípicos, se muestra de manera esquemática en este cuadro general. ARNm: ARN mensajero; miARN: microARN. Reproducido de Pasipoularides2, con permiso de International Journal of Cardiology y Elsevier.")
El genoma integra señales intrínsecas y ambientales. Los factores reguladores epigenéticos y ambientales bernardianos influyen a través del flujo de información (bidireccional) y las vías de señalización en la transición y expresión de los genes humanos, mientras que la cantidad de proteínas y sus funciones se ven afectadas por diversos ajustes postraduccionales. Dado que los estados epigenéticos son reversibles, pueden modificarse por factores ambientales que pueden contribuir a cambios modificables entre los fenotipos normales y anormales. s posible que la interferencia en la actividad de factores que modifican el estado de la cromatina afecte a la expresión de variantes génicas no deseadas. El organismo humano es de hecho un «supraorganismo», es decir, una mezcla de genes y rasgos humanos y del microbioma, incluido el metaboloma. La relación entre los diferentes componentes «-oma/-ómicos», la cantidad de proteínas, la modificación postraduccional y la localización y la actividad de las proteínas que afectan a los rasgos fenotípicos, se muestra de manera esquemática en este cuadro general. ARNm: ARN mensajero; miARN: microARN. Reproducido de Pasipoularides2, con permiso de International Journal of Cardiology y Elsevier.
Conviene introducir aquí una nota de precaución: los resultados de Pérez-Sánchez et al.1 indican que el ejercicio moderado puede no causar problemas, pero los ensayos clínicos, de discreto tamaño, no permiten confirmar que el ejercicio sea seguro en la MCH; para ello sería necesario aleatorizar a más de 2.000 pacientes y darles seguimiento durante ≈ 3 años. La conclusión de que un aumento de la actividad física no se asocia con la gravedad o el pronóstico de la enfermedad de los portadores de mutaciones causales deberá confirmarse en estudios prospectivos más amplios. En este contexto, convendrá investigar en profundidad el papel de la producción, la secreción y la expresión de citocinas/miocinas estimuladas por el ejercicio, ya que sus efectos beneficiosos cardiacos podrían proporcionar una explicación eficiente. De hecho, se sabe que las miocinas inducidas por el ejercicio tienen influencia en muchos órganos22. También en la MCH, las miocinas inducidas por el ejercicio podrían aportar efectos beneficiosos23, al estimular vías metabólicas, mejorar la captación de glucosa y aumentar el grado de oxidación de las grasas.
De hecho, los grandes ensayos de correlación entre genotipo/epigenotipo y fenotipo pueden revelar efectos de factores genéticos y ambientales poco conocidos que modulen el fenotipo de MCH, y la investigación básica paralela deberá esclarecer los mecanismos subyacentes. Las tensiones mecánicas, o su ausencia, actúan como factores ambientales/epigenéticos clave que afectan a los rasgos de MCH3,20,21. Estos factores modifican el panorama genómico al influir en las fuerzas moleculares que actúan; pueden incluir una ausencia anormal en la cavidad del VI estrechada de la MCH que se da con la tensión tangencial endocárdica/miocárdica normalmente intensa y fuerzas que «exprimen», ejercidas por el gran flujo rotatorio de llenado diastólico de los ventrículos derecho e izquierdo24–28.
NO SE TRATA SOLO DE LOS GENES, SINO DE LO QUE ESTÉN HACIENDOLos factores ambientales/epigenéticos pueden modificar el panorama de la MCH, ya que no es solo la secuencia del ADN lo que influye en lo que sucede. El ADN que forma nuestros genes no es capaz de tener una acción autónoma, por lo que su regulación es crucial. Aquello que el contexto génico (que engloba otros genes, además de factores ambientales/epigenéticos) induce a hacer a los genes de un individuo es tan importante como las variantes polimórficas presentes. Un gen anormal heredado no tiene ninguna consecuencia si los procesos epigenéticos lo mantienen inactivo. La epigenética incluye modificaciones reversibles y a veces heredables en nucleótidos o cromosomas que pueden modificar la expresión génica y la estructura de la cromatina sin alterar la secuencia primaria de nucleótidos del ADN (figura 3). Las modificaciones epigenéticas incluyen la metilación de ADN, la acetilación o metilación de las histonas y la arquitectura de la cromatina2,15,20,21,25–27. Estas modificaciones influyen en la regulación y expresión génica y pueden causar susceptibilidad o resistencia a diversas enfermedades.
Las histonas, que son proteínas macromoleculares relacionadas con el ensamblaje del ADN, se pueden modificar por factores epigenéticos a través de diversos procesos que modifican el acceso de la ARN polimerasa: la acetilación de las histonas hace que los segmentos de ADN vinculados pasen a ser más accesibles a la ARN polimerasa, lo cual conduce al inicio/aumento de la transcripción/expresión génica. Los grupos metilo se unen directamente a dinucleótidos CpG en los segmentos del ADN promotores de genes, generalmente para inhibir la transcripción, con lo que detienen/retardan la expresión génica.
Las vías epigenéticas actúan en la intersección entre los factores genéticos y ambientales (figura 3). Aunque no se sabe todavía de qué manera los factores ambientales alteran el ADN e influyen en la expresión génica a lo largo de la vida, la epigenética parece ser la interfaz de las interacciones entre genes y entorno (G×E) que modifican la expresión génica. Esta cuestión compleja se está estudiando desde el punto de vista energético en los ensayos clínicos en curso1. El perfil epigenético de un individuo se ve influido por factores tanto aleatorios como ambientales. Aunque los gemelos monocigotos son genéticamente idénticos, sus patrones de metilación del ADN van haciéndose más diferentes con el paso del tiempo. Los factores ambientales pueden tener efectos nocivos o protectores a través de procesos epigenéticos que pueden actuar en trastornos humanos como la MCH.
Descifrar las intrincadas cadenas de señalización que van de la proteína o las proteínas mutantes al fenotipo o los fenotipos clínicos e identificar los factores ambientales y epigenéticos que modifican la expresión de mutaciones/variantes polimórficas deberán generar nuevas perspectivas. Esto hace que el trabajo de Pérez-Sánchez et al.1 tenga interés en la búsqueda de un mejor conocimiento de la patogenia de la MCH y en la mejora de las intervenciones preventivas y terapéuticas.
CONCLUSIONESAunque se han hecho grandes progresos en el esclarecimiento de la patogenia de la MCH, los conocimientos actuales son insuficientes para predecir los fenotipos o los resultados clínicos de manera personalizada. Queda mucho por descubrir respecto a la penetrancia fenotípica y la posibilidad de una prevención o reversión del fenotipo cambiante. Los fenotipos de la enfermedad incluyen interacciones entre genes causales, genes modificadores y entorno. Los avances en la genómica funcional están pasando a ser cada vez más importantes en la investigación y la práctica clínica cardiológicas. No pasará mucho tiempo antes de que la creciente sofisticación genómica/epigenómica tenga la tan anunciada repercusión transformadora en el diagnóstico de la MCH, la estratificación del riesgo y la aplicación de medidas terapéuticas y preventivas a los pacientes, los portadores de mutaciones y las familias.
FINANCIACIÓNPara el trabajo del laboratorio de A. Pasipoularides que se cita aquí, se dispuso de ayuda para la investigación de: National Heart, Lung, and Blood Institute, subvención R01 HL 050446; National Science Foundation, subvención CDR 8622201, y North Carolina Supercomputing Center and Cray Research.
CONFLICTO DE INTERESESNo se declara ninguno.