
Se presenta el caso de una mujer de 27 años que ingresó en nuestro hospital por insuficiencia cardiaca. A la edad de 5 meses había ingresado por insuficiencia cardiaca aguda. Se realizó una ecocardiografía, que puso de manifiesto la existencia de una miocardiopatía dilatada con fracción de eyección del ventrículo izquierdo gravemente reducida, que después se recuperó completamente en el seguimiento, por lo que se consideró que estaba relacionada con una miocarditis aguda. La paciente presentaba nistagmo, distrofia de conos y bastones y atrofia de la vía óptica desde los primeros meses de vida, con ceguera bilateral y sordera perceptiva desde la adolescencia. Tenía antecedente de obesidad desde la infancia, y a los 14 años se le había diagnosticado diabetes mellitus tipo 2 e hipertrigliceridemia. Todos estos signos alertaban de una posible encefalopatía mitocondrial, pero la biopsia muscular y el estudio genético de Sanger fueron negativos para la mutación puntual A8344G involucrada en el síndrome de encefalomiopatía mitocondrial, acidosis láctica y episodios de tipo ictus (MELAS) y en la epilepsia monoclónica con fibras rojas rasgadas (MERRF).
En el ingreso actual, la paciente acudió por disnea en los últimos meses, que había progresado hasta alcanzar una clase funcional III/IV de la New York Heart Association.
La ecocardiografía transtorácica reveló una dilatación leve del ventrículo izquierdo con disfunción sistólica grave sin áreas de hipertrabeculación, hipertrofia o de disminución del espesor miocárdico tras la administración del contraste ecocardiográfico. La paciente evolucionó favorablemente con el tratamiento médico habitual, que resolvió los síntomas congestivos; recibió el alta y se le programó una resonancia magnética para completar la evaluación de la miocardiopatía dilatada.
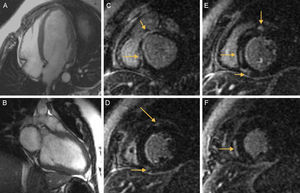
La resonancia magnética cardiaca confirmó un ventrículo izquierdo dilatado con disfunción sistólica grave, así como la existencia de captación patológica de contraste en la región subepicárdica, en el punto de inserción de ambos ventrículos, y captación intramiocárdica en el segmento septal medio, compatible con fibrosis miocárdica (figura A-F).
.")
A, B: imágenes de eco-gradiente de eje largo, tetracamerales y bicamerales en las que se observa un ventrículo izquierdo dilatado. C-F: captación tardía de gadolinio en la región subepicárdica en el punto de inserción de ambos ventrículos y captación de contraste intramiocárdica en el segmento septal medio (flechas).
A la vista de estas observaciones, y tras haber descartado, con alto grado de certeza según los estudios previos, una enfermedad mitocondrial, se estableció el diagnóstico de síndrome de Alström, puesto que la paciente cumplía los criterios clínicos diagnósticos (tabla). Se solicitó un estudio genético mediante una secuenciación de nueva generación para el gen ALMS1, que está implicado en esta enfermedad. Este estudio dio un resultado positivo para 2 mutaciones en los exones 8 y 11 del gen con relevancia patogénica, por lo que se estableció el diagnóstico definitivo.
Criterios diagnósticos de Marshall específicos para la edad
| Edad (años) | Criterios mayores | Criterios menores | Otros datos de apoyo | Diagnóstico |
|---|---|---|---|---|
| ≤ 2* | • Mutación ALMS 1 en 1 alelo y/o antecedentes familiares de SA • Vista (nistagmo, fotofobia) | • Obesidad • Miocardiopatía dilatada o insuficiencia cardiaca congestiva | • Infecciones pulmonares recurrentes • Dedos normales • Retraso en los hitos del desarrollo | 2 criterios mayores o 1 criterio mayor + 2 criterios menores |
| 3-14 | • Mutación ALMS 1 en 1 alelo y/o antecedentes familiares de SA • Vista (nistagmo, fotofobia, disminución de agudeza visual, distrofia de conos y bastones en el ERG) | • Obesidad y/o resistencia a la insulina • Antecedentes de miocardiopatía dilatada o insuficiencia cardiaca congestiva • Pérdida de audición • Disfunción hepática • Insuficiencia renal | • Infecciones pulmonares recurrentes • Dedos normales • Retraso en los hitos del desarrollo • Hiperlipemia • Escoliosis • Pies planos y anchos • Hipotiroidismo • Hipertensión • Déficit de GH • IU recurrente | 2 criterios mayores o 1 criterio mayor + 3 criterios menores |
| > 15 | • Mutación ALMS 1 en 1 alelo y/o antecedentes familiares de SA • Vista (ceguera legal, antecedentes de nistagmo en la primera o segunda infancia, distrofia de conos y bastones en el ERG) | • Obesidad y/o resistencia a la insulina y/o diabetes mellitus tipo 2 • Antecedentes de miocardiopatía dilatada o insuficiencia cardiaca congestiva • Pérdida de audición • Disfunción hepática • Insuficiencia renal • Baja estatura • Varones, hipogonadismo • Mujeres, menstruación irregular y/o hiperandrogenismo | • Infecciones pulmonares recurrentes • Dedos normales • Antecedentes de retraso del desarrollo • Hiperlipemia • Escoliosis • Pies planos y anchos • Hipotiroidismo • Hipertensión • Déficit de GH • Alopecia • IU recurrente o disfunción urinaria | 2 criterios mayores + 2 criterios menores o 1 criterio mayor + 4 criterios menores |
ERG: electrorretinograma; GH: hormona de crecimiento; IU: infecciones urinarias; SA: síndrome de Alström.
Esta paciente es el primer caso de esta enfermedad descrito en su familia. El estudio genético para la detección de esas mutaciones en sus padres fue negativo, por lo que al parecer se trata de una mutación espontánea en ese gen.
Hoy la paciente tiene 29 años y se encuentra en clase funcional I/IV de la New York Heart Association, sin episodios de descompensación, a pesar de que persistía una disfunción sistólica grave del ventrículo izquierdo en las visitas de seguimiento.
Alström et al.1 describieron el síndrome de Alström (SA) por primera vez en 1959. Tiene una prevalencia < 1/100.0001,2. El SA es una enfermedad genética de herencia autosómica recesiva que se caracteriza por una afección multisistémica producida por una mutación del gen ALMS1 situado en el cromosoma 2p133. Este gen codifica una proteína cuya mutación condiciona una fibrosis progresiva en diversos órganos y se caracteriza por distrofia de conos y bastones, pérdida de audición, obesidad central en la infancia, resistencia a la insulina e hiperinsulinemia, diabetes mellitus tipo 2, hipertrigliceridemia, baja talla en la edad adulta, miocardiopatía y disfunción renal, hepática y pulmonar progresivas. Los primeros síntomas aparecen durante la infancia y el desarrollo progresivo de una enfermedad multiorgánica reduce la esperanza de vida de estos pacientes2–4. Hasta el descubrimiento de las mutaciones del gen ALMS1, el diagnóstico se establecía basándose solo en el fenotipo. Sin embargo, el alto grado de variabilidad, incluso dentro de una misma familia, ocasiona dificultades para establecer una definición universal5. Marshall definió el SA mediante unos criterios específicos dependientes de la edad (tabla)6.
La afección ocular es uno de los signos cardinales del SA1,5,6 y da lugar a una disfunción visual progresiva hasta la ceguera, generalmente durante la segunda década de vida. Además, alrededor del 80% de los pacientes sufren una pérdida de audición neurosensitiva que progresa a lo largo de la vida4–6.
La obesidad infantil es una manifestación frecuente y temprana. Generalmente se acompaña de una expresión fenotípica característica, y a menudo contraen diabetes mellitus tipo 2 con resistencia a la insulina e hiperinsulinemia, así como hipertrigliceridemia. Debido a estas alteraciones, el perfil metabólico de estos pacientes lleva a un aumento del riesgo cardiovascular1,5.
El daño cardiaco se caracteriza por la aparición de una miocardiopatía dilatada con disfunción sistólica, fibrosis miocárdica y reducción de la masa miocárdica2,3. La afección cardiaca es una manifestación frecuente y la principal causa de morbilidad y mortalidad de estos pacientes, así como la primera causa de muerte en la infancia. Se puede manifestar como una insuficiencia cardiaca aguda en cualquier momento de la vida, si bien es frecuente que aparezca en las primeras semanas o meses de vida como primera manifestación, como en nuestro caso2–6. La posterior recuperación de la función cardiaca es frecuente, como lo es también la recurrencia durante la adolescencia o en la edad adulta2,5.
El pronóstico es variable y depende de cómo evolucionen las afecciones de los diferentes órganos y sistemas. Generalmente, la esperanza de vida es de menos de 50 años5. Aunque no hay ningún tratamiento específico y las medidas adoptadas deben dirigirse a tratar el daño de cada órgano afectado, un diagnóstico precoz, un abordaje multidisciplinario y las estrategias de prevención adecuadas permiten retardar la progresión y con ello mejorar la supervivencia de estos pacientes2,3,5.
Es frecuente que los síndromes genéticos sean difíciles de diagnosticar y, en la mayoría de los casos, carecen de un tratamiento específico. Un alto grado de sospecha es crucial para establecer el diagnóstico correcto, lo cual no solo permite mejorar la asistencia de los pacientes, sino que es necesario para proporcionar un asesoramiento genético adecuado y orientar el estudio familiar, si estuviera indicado.