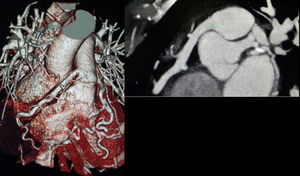
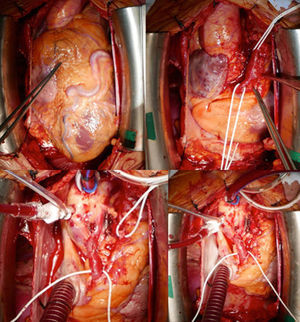
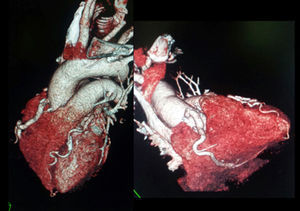
Mujer de 52 años, hipertensa como factor de riesgo cardiovascular. Presenta historia de dolores torácicos atípicos, aunque últimamente presenta opresión precordial al realizar esfuerzos moderados. En la exploración física destaca soplo sistólico II/IV. Se realiza ecocardiografía transtorácica con ventrículo izquierdo límite (diámetro telediastólico, 52mm) y función sistólica global/segmentaria conservadas; ergometría (protocolo de Bruce) clínicamente positiva (opresión precordial) y eléctricamente no concluyente. Se solicita tomografía computarizada coronaria que muestra nacimiento anómalo de la arteria coronaria derecha (CD), dilatada, desde la arteria pulmonar (AP), con nacimiento normal de tronco coronario y la arteria descendente anterior (DA) muy dilatada (figura 1). Se diagnostica de síndrome de ARCAPA (anomalous origin of the right coronary artery from the pulmonary artery) y se decide intervenir quirúrgicamente. Se observa CD y DA muy dilatadas. Se realiza disección de CD cerca del origen de la AP y anastomosis de coronaria anómala en aorta con resolución del shunt izquierda-derecha (figura 2). En la tomografía computarizada de control (figura 3) muestra buena evolución posterior.



El origen anómalo de las arterias coronarias en la AP es infrecuente, con menor incidencia el de CD (síndrome de ARCAPA). Corresponde al 0,002% de todas las cardiopatías congénitas y se diagnostica con mayor frecuencia en la edad adulta. Se asocia a aneurismas de arterias coronarias, AP y trayecto fistuloso. En la mayoría de casos el tratamiento es quirúrgico (reimplante de arteria coronaria anómala en aorta). Aunque existe controversia en el tratamiento que hay que realizar en pacientes asintomáticos, la mayoría de autores está a favor de la corrección quirúrgica; ya que los pacientes tendrían aumento progresivo del shunt izquierda-derecha y pobre reserva coronaria y estarían predispuestos a isquemia, arritmias y muerte súbita.
Full English text available from: www.revespcardiol.org/en