La miocardiopatía dilatada (MCD) idiopática tiene una causa genética en hasta un 20% de los casos1. Los exámenes clínicos de detección sistemática familiares revelan que un 20-48% de los casos iniciales (probandos) tienen familiares afectados, lo cual concuerda con un diagnóstico de MCD familiar1,2.
El síndrome de Barth es un trastorno recesivo ligado al cromosoma X causado por mutaciones del gen de tafazzina (TAZ)3. Se caracteriza por MCD, neutropenia y aciduria 3-metilglutacónica4 y la esperanza de vida es limitada durante la fase inicial de la infancia.
Presentamos el curso clínico de un varón de 30 años con síndrome de Barth y los resultados del estudio genético del gen TAZ en sus familiares. Con objeto de evaluar la intervención del gen TAZ en la etiología de la MCD y el ventrículo izquierdo no compactado, estudiamos el gen TAZ en 48 pacientes con MCD y ventrículo izquierdo no compactado.
Nuestro paciente fue evaluado por primera vez por una infección respiratoria en el departamento de pediatría cuando tenía 10 meses de edad. En ese momento se le diagnosticó cardiomegalia, disfunción sistólica y miopatía. El desarrollo del paciente fue normal. Durante la fase inicial de la infancia sufrió frecuentes infecciones. A los 20 años, los síntomas principales consistían en fatiga y claudicación muscular. Pesaba 71 kg y medía 180 cm de estatura.
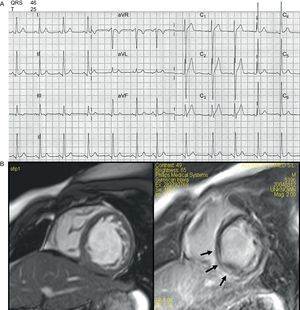
Desde el punto de vista cardiológico, nunca había tenido síncopes y sufría episodios ocasionales de dolor torácico y palpitaciones. Un electrocardiograma (ECG) reciente había sido normal en esencia (fig. 1A), con ritmo sinusal y un intervalo PR ligeramente corto (0,12 ms), y con un QRS de voltaje levemente elevado, en especial con ondas S profundas en V1-V2 y elevación del ST con un patrón de repolarización temprana. Los intervalos QRS y QT eran normales. Los registros Holter ambulatorios de 24 h y de 7 días realizados repetidamente no habían podido identificar ninguna arritmia aparte de la taquicardia sinusal. Se evaluó la capacidad de ejercicio del paciente en cinta sin fin, en la que sólo pudo realizar ejercicio durante 4 min. La frecuencia cardiaca durante la prueba pasó de 78 a 188 lpm (el 96% del valor esperado), y se alcanzó un valor máximo de VO2 de 13,3ml/kg/min. La prueba se interrumpió por disnea. No hubo ningún cambio del ST-T y no se produjeron arritmias durante la prueba cardiorrespiratoria. Se añadió entonces una dosis baja de bloqueadores beta al tratamiento crónico de losartán, que se toleró bien y produjo un efecto beneficioso en los síntomas.

Una ecocardiografía reciente había mostrado un deterioro leve de la función sistólica ventricular izquierda (la fracción de eyección era del 50%) con una dimensión telesistólica normal (50 mm). La resonancia magnética confirmó los resultados ecocardiográficos y mostró unos segmentos medios y apicales del ventrículo izquierdo hipertrabeculados, que cumplían los criterios de no compactado. Se observó una línea de captación de contraste de gadolinio en la pared inferoposterior (miocardio medio) (fig. 1B).
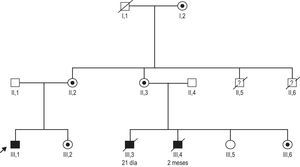
En los antecedentes familiares destacaban dos primos varones fallecidos por insuficiencia cardiaca (a las edades de 1 y 2 meses) y dos tíos varones que habían muerto también en la primera infancia con sospecha de enfermedad infecciosa (fig. 2). Los exámenes hematológicos y metabólicos mostraron que el paciente tenía neutropenia, acidemia láctica y aciduria 3-metilglutacónica; se sospechó un diagnóstico de síndrome de Barth.

Se identificó una mutación de c.280C>T (R94C)6 en el gen TAZ de nuestro paciente (III,1) (fig. 2). Este cambio estaba presente también en el primo varón de 2 meses que falleció por insuficiencia cardiaca (III,6). No se dispuso de muestras de los demás familiares varones con sospecha de que podrían estar afectados. La madre de nuestro paciente (II,2), una tía (II,3), una hermana (III,2) y la abuela (I,2) eran portadoras de la mutación. Se evaluó a todas las mujeres portadoras, y las exploraciones cardiacas fueron normales. El espectro clínico en las portadoras de esta mutación es amplio; la relación genotipo-fenotipo se conoce sólo de manera incompleta, y en esta familia había 4 bebés varones que fallecieron por insuficiencia cardiaca o una enfermedad infecciosa. Hay otros factores genéticos o ambientales que pueden haber desempeñado un papel en el curso poco habitual de la enfermedad de nuestro paciente inicial.
Se examinó también a un total de 48 pacientes consecutivos pediátricos (n = 17; edad, 2 días-14 años; 12 varones) y adultos (n = 31; edad, 15-72 años; 24 varones) con MCD idiopática o ventrículo izquierdo no compactado. La media de la fracción de eyección ventricular izquierda fue del 41,6% ± 19,7%. De ellos, 22 tenían ventrículo izquierdo no compactado (11, ventrículo izquierdo no compactado solamente); 7 se encontraban en la clase IV de la New York Heart Association, 8 estaban en clase III y 11 en clase II. Hubo 3 trasplantes de corazón en este grupo. El estudio genético realizado en estos pacientes no ha mostrado mutaciones del gen TAZ.
Sólo hay otro caso similar de síndrome de Barth en un adulto (de 35 años), descrito por Kelley et al5 (1991). Los registros Holter repetidos no han mostrado arritmia o trastorno de la conducción en nuestro paciente. El diagnóstico genético es esencial para el consejo genético en este trastorno ligado al cromosoma X. Un diagnóstico precoz y exacto puede ser útil para el tratamiento médico y para mejorar el pronóstico.
A pesar del mal pronóstico del síndrome de Barth en el lactante, los pacientes pueden sobrevivir hasta la edad adulta. No hemos encontrado mutaciones del gen TAZ en nuestra cohorte de pacientes pediátricos y adultos con MCD aislada o ventrículo izquierdo no compactado, por lo que se debe sospechar síndrome de Barth si hay antecedentes familiares con varones que hayan fallecido precozmente por una MCD, así como en varones adultos con datos clínicos característicos, puesto que nuestro caso indica que es posible sobrevivir al síndrome.
FINANCIACIÓNEste estudio se ha financiado con una subvención de investigación de la Fundación Española del Corazón-Sociedad Española de Cardiología 2007 y por la Red de Investigación Cardiovascular (RECAVA).