El síndrome LEOPARD es una enfermedad autosómica dominante relacionada con el síndrome de Noonan, aunque menos conocida. El objetivo del presente estudio es describir las características clínicas y moleculares de una serie amplia de pacientes con síndrome LEOPARD.
MétodosSe obtuvieron datos clínicos de 19 pacientes procedentes de 10 hospitales. Se estudiaron los genes PTPN11, RAF1 y BRAF mediante secuenciación bidireccional de los exones más recurrentes.
ResultadosTras las dismorfias faciales, la principal característica descrita es la cardiopatía congénita (88%). La más frecuente es la miocardiopatía hipertrófica (71%), por delante de la estenosis pulmonar (35%). Se describió lentiginosis múltiple o manchas café con leche en un 84% y sordera en 3 pacientes; 16 pacientes (84%) portaban mutación en PTPN11 (en 10 de ellos, la mutación recurrente en el síndrome LEOPARD, p.Thr468Met) (NP_002825.3). En otros 2 pacientes se identificó mutación en RAF1 y 1 solo en BRAF. En comparación con otros síndromes neurocardiofaciocutáneos, los pacientes con LEOPARD tienen mayor prevalencia de miocardiopatía hipertrófica y lesiones cutáneas y menor prevalencia de estenosis pulmonar y talla baja.
ConclusionesEl síndrome LEOPARD presenta algunas características distintivas además de la lentiginosis múltiple, como son la mayor frecuencia de miocardiopatia hipertrófica y menor prevalencia de talla baja. Dadas las potenciales implicaciones clínicas de la miocardiopatía hipertrófica, se debe buscar activamente en los pacientes del espectro clínico del síndrome de Noonan, y muy especialmente en aquellos con síndrome LEOPARD.
Palabras clave
El síndrome LEOPARD (SL), o síndrome de Noonan (SN) con lentiginosis múltiple (OMIM 151100) es una enfermedad autosómica dominante caracterizada por lentiginosis múltiple o manchas café con leche, alteraciones electrocardiográficas, hipertelorismo ocular, estenosis pulmonar valvular o miocardiopatía hipertrófica, anomalías genitales, retraso constitucional y sordera (deafness)1. El SL comparte muchas características con el SN (OMIM 163950), que se caracteriza por la asociación de cardiopatía congénita, talla baja y malformaciones craneofaciales2 y habitualmente no incluye la lentiginosis múltiple ni la sordera entre sus manifestaciones. Se han identificado mutaciones en el gen PTPN11 en un 50% de los casos de SN3–5 y en un 85% de los casos de SL6,7. En este también se han identificado mutaciones en los genes RAF18 y BRAF9.
Aunque no hay datos exactos acerca de su prevalencia, se piensa que el SN está presente en 1/1.000-2.500 recién nacidos vivos, lo que hace suponer que se trata de un trastorno infradiagnosticado10. El SL es menos frecuente, y se desconoce su prevalencia exacta al nacimiento. Hasta la fecha se han descrito al menos 200 casos en la literatura médica, y recientemente se ha publicado una revisión amplia de esta entidad11. Ambos trastornos tienen una amplia variabilidad fenotípica, lo que dificulta su identificación y correcto diagnóstico y hace del estudio genético una herramienta útil para el diagnóstico diferencial. Asimismo, la dificultad en el diagnóstico estriba en el carácter evolutivo y cambiante de muchas de sus características, lo que hace de la cardiopatía congénita un dato objetivable muy valioso a la hora de identificar el síndrome.
Presentamos la descripción fenotípica de una serie de pacientes con diagnóstico de SL, caracterizados mediante estudio genético mutacional de los genes PTPN11, RAF1 y BRAF, y su comparación estadística con una serie amplia de pacientes con otros síndromes neurocardiofaciocutáneos (SNCFC) a su vez caracterizados mediante estudio genético mutacional de los genes PTPN11, SOS1, RAF1, BRAF y HRAS.
MÉTODOSEvaluación clínicaLos pacientes fueron evaluados por genetistas clínicos, cardiopediatras o endocrinopediatras involucrados en un estudio multicéntrico de relación genotipo-fenotipo desarrollado en el territorio nacional, y fueron diagnosticados de SL según los criterios diagnósticos descritos por Voron at al12. Las muestras de sangre o ADN de los pacientes nos fueron remitidos desde los hospitales solicitantes por los clínicos responsables, que obtuvieron los consentimientos informados. Se realizó una primera aproximación clínica basada en el formulario preanalítico descrito en Ezquieta et al13 para la selección de casos. Utilizando una base de datos Access consensuada entre los clínicos implicados, se recabó información de las características clínicas referentes a cardiopatía congénita, alteraciones cutáneas, desarrollo ponderoestatural, problemas de audición, anomalías genitales y características faciales de los pacientes. Su fenotipo facial se clasificó como típico cuando había tres o más de las siguientes malformaciones: hipertelorismo ocular, ptosis palpebral, orejas de implantación baja e inclinación de las hendiduras palpebrales hacia abajo, y como sugestivo cuando no cumplían este criterio. La talla se evaluó en desviaciones estándar respecto a la población de referencia14, y se consideró baja cuando era <–2 desviaciones estándar. A todos los pacientes se les realizó electrocardiograma y ecocardiograma valorados por un cardiólogo infantil. La estenosis pulmonar valvular se diagnosticó por criterios ecográficos clásicos. Se diagnosticó miocardiopatía hipertrófica cuando el grosor de la pared anterior del ventrículo izquierdo era >2 desviaciones estándar para la edad.
En 2 casos con diagnóstico de SN en los que el estudio genético identificó una mutación típica de LEOPARD, se controló al paciente evolutivamente, y se pudo constatar la aparición de la lentiginosis múltiple, con lo que se modificó el diagnóstico (casos 1 y 8). En cuanto a los casos familiares, una familia de 3 miembros fue evaluada clínicamente desde el inicio (familia A), y el estudio genético posterior confirmó la sospecha. En el otro caso familiar evaluado (familia B), fue el estudio molecular lo que identificó a la madre del caso índice como portadora de la mutación, y ello motivó su posterior estudio clínico.
En cuanto al grupo de pacientes con otros SNCFC, se utilizaron los datos provisionales del mismo estudio multicéntrico de correlación genotipo-fenotipo, actualmente en curso13,15. Para los pacientes con SN se emplearon los criterios diagnósticos de Van der Burgt10; en aquellos con síndrome cardiofaciocutáneo, el índice cardiofaciocutáneo16, y en los casos de sospecha clínica de síndrome de Costello, se consideró confirmado el síndrome si se identificaba mutación en HRAS17. De todos estos pacientes, se recogieron los datos clínicos con la misma metodología descrita para los pacientes con SL.
Análisis mutacionalSe obtuvieron muestras de sangre de los pacientes y sus familiares previo consentimiento informado. Se extrajo ADN genómico por los procedimientos habituales. Se procedió a amplificación mediante reacción en cadena de la polimerasa (PCR) utilizando los cebadores y los parámetros de ciclado descritos originalmente por Tartaglia et al18. Posteriormente se procedió a secuenciación bidireccional de las regiones codificantes y regiones intrónicas adyacentes con un ABI Prism Genotyper®. Se empleó el software SeqScape 2.5 para el análisis de los electroforetogramas obtenidos.
Análisis estadísticoEl análisis estadístico se llevó a cabo usando el test exacto de Fisher. Se consideró estadísticamente significativo p<0,05. Para el análisis se empleó el paquete estadístico SPSS 19.0.
RESULTADOSEl estudio incluyó a 19 pacientes, 13 varones y 6 mujeres (edad al diagnóstico, 11 meses-49 años; media de edad, 7,4 años). Los casos fueron remitidos desde nueve hospitales de cinco comunidades españolas y un hospital de Belgrado (Serbia). Los casos fueron identificados como familiares en 5 pacientes (familias A y B; el 26% de los casos; intervalo de confianza del 95% [IC95%], 9,1-51,2%), y se consideró esporádicos al resto. La tabla 1 resume los hallazgos fenotípicos y moleculares. Del total de los casos, 15 cumplían los criterios clínicos de Voron, mientras que en otros 4 se consideró que tenían una «forma parcial de SL». Estos pacientes presentaban características clínicas típicas del SL, pero no tenían lentiginosis múltiple ni historia familiar de síndrome de SL. Los 4 eran portadores de la mutación en PTPN11 típica de SL, p.Thr468Met, y estaban en edad pediátrica en el momento de la evaluación, por lo que aún no se puede asegurar que no vayan a sufrir las manifestaciones cutáneas características del síndrome con el crecimiento. Las anomalías craneofaciales (el 100% incluyendo fenotipos típico y sugestivo; IC95%, 82,4-100%) y cardiacas (88%; IC95%, 63,6-98,5%) y las lesiones cutáneas (84%; IC95%, 60,4-96,6%) fueron las características más frecuentemente descritas, seguidas por la talla baja (37%; IC95%, 16,3-61,6%) y las anomalías genitales (criptorquidia en 3 pacientes, el 16% del total, el 23% de los varones; IC95%, 5-53,8%). En 3 pacientes se diagnosticó hipoacusia, aunque sólo 1 de ellos (5%; IC95%, 0,1-26%) presentaba componente neurosensorial; 7 pacientes presentaron retraso psicomotor (37%; IC95%, 16,3-61,6%), todos ellos de carácter leve. Los 15 pacientes con fenotipo completo presentaban lentiginosis múltiple (79%, IC95%, 54,4-93,9%) y 11 de ellos tenían además manchas café con leche (58%; IC95%, 33,5-79,7%). Uno de los pacientes con fenotipo parcial presentaba también manchas café con leche, mientras que los otros 3, con edades comprendidas entre los 10 meses y los 14 años, aún no tenían lesiones cutáneas. Dos pacientes no emparentados que en la valoración inicial no tenían manifestaciones cutáneas, el número 1 y el número 8, con las mutaciones p.Thr468Met y p.Gln510Arg respectivamente, desarrollaron en el seguimiento posterior la lentiginosis múltiple que caracteriza el síndrome, lo que ilustra el carácter evolutivo de sus manifestaciones.
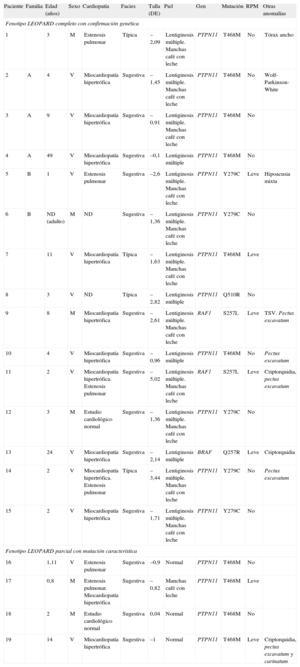
Características clínicas y moleculares de 19 pacientes con síndrome LEOPARD
| Paciente | Familia | Edad (años) | Sexo | Cardiopatía | Facies | Talla (DE) | Piel | Gen | Mutación | RPM | Otras anomalías |
| Fenotipo LEOPARD completo con confirmación genética | |||||||||||
| 1 | 3 | M | Estenosis pulmonar | Típica | –2,09 | Lentiginosis múltiple. Manchas café con leche | PTPN11 | T468M | No | Tórax ancho | |
| 2 | A | 4 | V | Miocardiopatía hipertrófica | Sugestiva | –1,45 | Lentiginosis múltiple. Manchas café con leche | PTPN11 | T468M | No | Wolf-Parkinson-White |
| 3 | A | 9 | V | Miocardiopatía hipertrófica | Sugestiva | –0,91 | Lentiginosis múltiple. Manchas café con leche | PTPN11 | T468M | No | |
| 4 | A | 49 | V | Miocardiopatía hipertrófica | Sugestiva | –0,1 | Lentiginosis múltiple | PTPN11 | T468M | No | |
| 5 | B | 1 | V | Estenosis pulmonar | Sugestiva | –2,6 | Lentiginosis múltiple. Manchas café con leche | PTPN11 | Y279C | Leve | Hipoacusia mixta |
| 6 | B | ND (adulto) | M | ND | Sugestiva | –1,36 | Lentiginosis múltiple. Manchas café con leche | PTPN11 | Y279C | No | |
| 7 | 11 | V | Miocardiopatía hipertrófica | Típica | –1,63 | Lentiginosis múltiple. Manchas café con leche | PTPN11 | T468M | Leve | ||
| 8 | 3 | V | ND | Típica | –2,82 | Lentiginosis múltiple | PTPN11 | Q510R | No | ||
| 9 | 8 | M | Miocardiopatía hipertrófica | Sugestiva | –2,61 | Lentiginosis múltiple. Manchas café con leche | RAF1 | S257L | Leve | TSV. Pectus excavatum | |
| 10 | 4 | V | Miocardiopatía hipertrófica | Sugestiva | –0,96 | Lentiginosis múltiple | PTPN11 | T468M | No | Pectus excavatum | |
| 11 | 2 | V | Miocardiopatía hipertrófica. Estenosis pulmonar | Sugestiva | –5,02 | Lentiginosis múltiple. Manchas café con leche | RAF1 | S257L | Leve | Criptorquidia, pectus excavatum | |
| 12 | 3 | M | Estudio cardiológico normal | Sugestiva | –1,36 | Lentiginosis múltiple. Manchas café con leche | PTPN11 | Y279C | No | ||
| 13 | 24 | V | Miocardiopatía hipertrófica | Sugestiva | –2,14 | Lentiginosis múltiple | BRAF | Q257R | Leve | Criptorquidia | |
| 14 | 2 | V | Miocardiopatía hipertrófica. Estenosis pulmonar | Típica | –3,44 | Lentiginosis múltiple. Manchas café con leche | PTPN11 | Y279C | No | Pectus excavatum | |
| 15 | 2 | V | Miocardiopatía hipertrófica | Sugestiva | –1,71 | Lentiginosis múltiple. Manchas café con leche | PTPN11 | Y279C | No | ||
| Fenotipo LEOPARD parcial con mutación característica | |||||||||||
| 16 | 1,11 | V | Estenosis pulmonar | Sugestiva | –0,9 | Normal | PTPN11 | T468M | No | ||
| 17 | 0,8 | M | Estenosis pulmonar. Miocardiopatía hipertrófica | Sugestiva | –0,82 | Manchas café con leche | PTPN11 | T468M | Leve | ||
| 18 | 2 | M | Estudio cardiológico normal | Sugestiva | 0,04 | Normal | PTPN11 | T468M | No | ||
| 19 | 14 | V | Miocardiopatía hipertrófica | Sugestiva | –1 | Normal | PTPN11 | T468M | Leve | Criptorquidia, pectus excavatum y carinatum | |
DE: desviaciones estándar; M: mujer; ND: no disponible; RPM: retraso psicomotor; TSV: taquicardias supraventriculares; V: varón.
Los casos familiares están marcados con letras (A y B). Los casos 2 y 5 son los casos índice de las familias A y B respectivamente.
La cardiopatía congénita más frecuente fue la miocardiopatía hipertrófica (71%; IC95%, 44-89,7%), seguida de la estenosis pulmonar valvular (35%; IC95%, 14,2-61,7%). Tres pacientes (18%; IC95%, 3,8-43,4%) presentaban ambos trastornos. La única cardiopatía congénita diferente de estas fue la coartación de aorta en un paciente que además tenía miocardiopatía hipertrófica. En este paciente la coartación se resolvió sin necesidad de cirugía correctora, lo que pondría en entredicho el diagnóstico de miocardiopatía hipertrófica, si bien la miocardiopatía persistió varios años después de resuelta la coartación. Sólo 2 casos (12%; IC95%, 1,5-36,4%) de los evaluados tenían una valoración cardiológica normal. Uno de los pacientes fue diagnosticado a los 2 meses de vida de un síndrome de Wolf-Parkinson-White, a raíz de un episodio de taquicardia supraventricular, y otro paciente padecía también taquicardias supraventriculares. Ningún paciente sufrió muerte súbita ni precisó implantación de desfibrilador.
El análisis molecular de los genes PTPN11, RAF1 y BRAF detectó mutación en los 19 pacientes. La mutación más frecuente fue p.Thr468Met (10 pacientes, 53%; IC95%, 28,9-75,6%). En 2 pacientes relacionados (madre e hijo, familia B) se identificó el cambio aminoacídico p.Tyr279Cys, así como en otros 3 pacientes no emparentados (en total un 26% de la serie; IC9%, 9,2-51,2%) y en otros 2 no emparentados, la variante p.Ser257Leu en RAF1. Los pacientes con mutación en RAF1 presentaban miocardiopatía hipertrófica y algunas de las tallas más bajas de nuestra serie. Finalmente, en 1 paciente se detectó un cambio de secuencia en el gen BRAF.
La comparación entre pacientes con SL y nuestra serie de pacientes con otros SNCFC incluyó a 87 con SN por mutación en PTPN11 (68 pacientes, 78%), SOS1 (15, 17%) y RAF1 (4, 5%), 5 con síndrome cardiofaciocutáneo con mutación en BRAF, y 1 paciente con síndrome de Costello por mutación en HRAS. Los pacientes procedían de 31 hospitales de 11 comunidades autónomas españolas, un hospital de Belgrado (Serbia) y un hospital de Buenos Aires (Argentina).
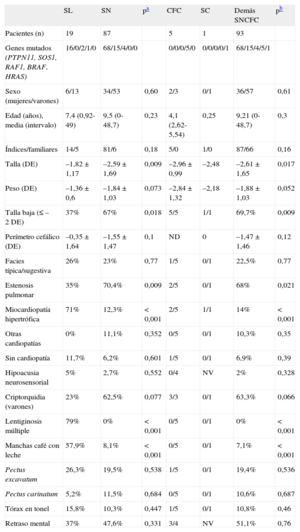
La tabla 2 reúne las características clínicas de cada uno de estos grupos de pacientes y de todos ellos en comparación con el SL. El SL se relaciona con la presencia de lentiginosis múltiple y de manchas café con leche (p<0,001). De igual forma, se evidencia una fuerte asociación entre el SL y la miocardiopatía hipertrófica, así como una talla más alta (p<0,05).
Comparación de algunos rasgos fenotípicos entre pacientes con síndrome LEOPARD y pacientes con otros síndromes neurocardiofaciocutáneos
| SL | SN | pa | CFC | SC | Demás SNCFC | pb | |
| Pacientes (n) | 19 | 87 | 5 | 1 | 93 | ||
| Genes mutados (PTPN11, SOS1, RAF1, BRAF, HRAS) | 16/0/2/1/0 | 68/15/4/0/0 | 0/0/0/5/0 | 0/0/0/0/1 | 68/15/4/5/1 | ||
| Sexo (mujeres/varones) | 6/13 | 34/53 | 0,60 | 2/3 | 0/1 | 36/57 | 0,61 |
| Edad (años), media (intervalo) | 7,4 (0,92-49) | 9,5 (0-48,7) | 0,23 | 4,1 (2,62-5,54) | 0,25 | 9,21 (0-48,7) | 0,3 |
| Índices/familiares | 14/5 | 81/6 | 0,18 | 5/0 | 1/0 | 87/66 | 0,16 |
| Talla (DE) | –1,82±1,17 | –2,59±1,69 | 0,009 | –2,96±0,99 | –2,48 | –2,61±1,65 | 0,017 |
| Peso (DE) | –1,36±0,6 | –1,84±1,03 | 0,073 | –2,84±1,32 | –2,18 | –1,88±1,03 | 0,052 |
| Talla baja (≤–2 DE) | 37% | 67% | 0,018 | 5/5 | 1/1 | 69,7% | 0,009 |
| Perímetro cefálico (DE) | –0,35±1,64 | –1,55±1,47 | 0,1 | ND | 0 | –1,47±1,46 | 0,12 |
| Facies típica/sugestiva | 26% | 23% | 0,77 | 1/5 | 0/1 | 22,5% | 0,77 |
| Estenosis pulmonar | 35% | 70,4% | 0,009 | 2/5 | 0/1 | 68% | 0,021 |
| Miocardiopatía hipertrófica | 71% | 12,3% | <0,001 | 2/5 | 1/1 | 14% | <0,001 |
| Otras cardiopatías | 0% | 11,1% | 0,352 | 0/5 | 0/1 | 10,3% | 0,35 |
| Sin cardiopatía | 11,7% | 6,2% | 0,601 | 1/5 | 0/1 | 6,9% | 0,39 |
| Hipoacusia neurosensorial | 5% | 2,7% | 0,552 | 0/4 | NV | 2% | 0,328 |
| Criptorquidia (varones) | 23% | 62,5% | 0,077 | 3/3 | 0/1 | 63,3% | 0,066 |
| Lentiginosis múltiple | 79% | 0% | <0,001 | 0/5 | 0/1 | 0% | <0,001 |
| Manchas café con leche | 57,9% | 8,1% | <0,001 | 0/5 | 0/1 | 7,1% | <0,001 |
| Pectus excavatum | 26,3% | 19,5% | 0,538 | 1/5 | 0/1 | 19,4% | 0,536 |
| Pectus carinatum | 5,2% | 11,5% | 0,684 | 0/5 | 0/1 | 10,6% | 0,687 |
| Tórax en tonel | 15,8% | 10,3% | 0,447 | 1/5 | 0/1 | 10,8% | 0,46 |
| Retraso mental | 37% | 47,6% | 0,331 | 3/4 | NV | 51,1% | 0,76 |
DE: desviaciones estándar; ND: no disponible; NV: no valorable; SC: síndrome de Costello; SCFC: síndrome cardiofaciocutáneo; SL: síndrome LEOPARD; SN: síndrome de Noonan; SNCFC: síndromes neurocardiofaciocutáneos.
SL: 15 pacientes con fenotipo LEOPARD completo y 4 pacientes con fenotipo LEOPARD parcial y mutación T468M en PTPN11 (datos expresados en porcentaje).
SN: 87 pacientes con SN confirmado genéticamente.
SCFC: 5 pacientes con SCFC confirmado genéticamente (datos expresados en cocientes).
SC: 1 caso de SC confirmado genéticamente (datos expresados en cocientes).
Demás SNCFC: pacientes con SNCFC distintos de LEOPARD (SN + SCFC + SC; datos expresados en porcentaje).
Como han apuntado algunos autores, una clasificación de las miocardiopatías apoyada en su base molecular contribuye a un manejo más apropiado19. En el caso de la miocardiopatía hipertrófica, se debe recordar el SN y el SL como algunas de las entidades que considerar20, máxime cuando el fenotipo acompañante puede variar desde casos letales a pacientes completamente asintomáticos desde el punto de vista cardiológico. Sólo un alto índice de sospecha permitirá al clínico considerar esta opción diagnóstica en los casos poco expresivos.
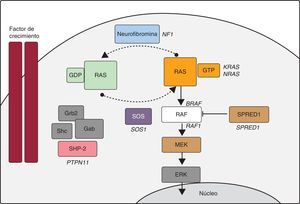
En los últimos años, los avances de la biología molecular han permitido dilucidar muchos aspectos de la etiología del SN y otras entidades con fenotipos solapantes (SL, síndrome de Costello, síndrome cardiofaciocutáneo, neurofibromatosis tipo 1 y síndrome de Legius). Algunos autores han decidido agrupar estos trastornos bajo la denominación de SNCFC21, síndromes RAS-MAPK o rasopatías, dado que parece que en la base de todos ellos reside un trastorno de la vía de señalización intracelular RAS-MAPK (figura). En el SN se han identificado mutaciones en el gen PTPN11 en aproximadamente un 50% de los pacientes, y se han descrito mutaciones en otros genes de la vía RAS-MAPK (SOS122, RAF18, KRAS23, MAP2K124, BRAF9, NRAS25 y SHOC226). Todavía hoy cerca de un 30% de los pacientes diagnosticados de SN no tiene una causa genética definida.
, MEK (MEK1, MEK2) y, por último, ERK.")
Cascada RAS-MAPK. La unión de un factor de crecimiento a un receptor de tirosincinasa activa efectores intracelulares como SHP2, que a su vez reclutan intercambiadores de guaninas como SOS1, que promueven el intercambio GDP/GTP en las proteínas RAS, las cuales se activan por fosforilación. RAS-GTP activa consecuentemente las distintas isoformas de RAF (RAF1, BRAF), MEK (MEK1, MEK2) y, por último, ERK.
En cuanto al SL, cerca de un 85% de los pacientes tienen mutaciones en el gen PTPN11. Se han descrito 11 mutaciones diferentes en PTPN11, pero el 65% de ellas se corresponden con dos altamente recurrentes: p.Tyr279Cys y p.Thr468Met7. Aproximadamente en un tercio de los pacientes PTPN11 negativos se han identificado mutaciones en RAF18, otro gen de la cascada RAS-MAPK. Finalmente, se han descrito mutaciones en BRAF9,27, un gen cuyas mutaciones se habían asociado fundamentalmente al síndrome cardiofaciocutáneo.
En las últimas décadas, el estudio molecular se ha demostrado como una herramienta útil en el diagnóstico diferencial de estas entidades solapantes, como ilustran varios casos de nuestra serie. Entre nuestros pacientes, la mutación más frecuente en PTPN11 es p.Thr468Met, seguida de p.Tyr279Cys. El seguimiento evolutivo de pacientes con la mutación p.Thr468Met ha permitido reorientar el diagnóstico en 2 de ellos al aparecer evolutivamente la lentiginosis múltiple, motivo por el que, como otros autores7, hemos considerado fenotipos parciales de SL a los pacientes con SN por p.Thr468Met en edad pediátrica. Las mutaciones con cambio en el aminoácido Gln510 se han asociado tanto a SN como a SL28, y en nuestra serie tenemos pacientes con cambios de secuencia que predicen el cambio de ese aminoácido por otros residuos (p.Gln510Glu, p.Gln510Pro) con diagnóstico de SN y edad adulta (por lo que no es previsible que el diagnóstico cambie y, por lo tanto, no se lo ha incluido en este estudio). Por otro lado, con la variante p.Gln510Arg, el paciente 8 desarrolló lentiginosis durante el seguimiento clínico en edad pediátrica, y se modificó el diagnóstico. En cuanto a RAF1, se identificó la mutación p.Ser257Leu en 2 casos. En 1 paciente negativo para PTPN11 y RAF1 se identificó mutación en BRAF, lo que demuestra la pertinencia de estudiar este gen en los pacientes con SL. Nuestro estudio confirma el alto rendimiento del estudio genético en pacientes con SL, su estrecha asociación con el gen PTPN11 y, en menor medida, RAF1 y BRAF.
Las claves diagnósticas de este síndrome son las manifestaciones cutáneas, que en ocasiones no se desarrollan completamente hasta pasada la pubertad29,30, la sordera, que no es tan frecuente como en las descripciones iniciales del síndrome, y la cardiopatía congénita. Si bien inicialmente se describió la estenosis pulmonar valvular como la más frecuente de las cardiopatías encontradas, actualmente se identifica con más frecuencia la miocardiopatía hipertrófica7,11,31, y en nuestra serie esta cardiopatía congénita se asocia al SL con significación estadística cuando lo comparamos con otros SNCFC. En nuestro medio, la cardiopatía congénita es la alteración que ha conducido al diagnóstico definitivo en una gran proporción de los casos, y sigue siendo la comorbilidad más peligrosa para estos pacientes32. La estenosis pulmonar valvular se identificó en 6 de nuestros 19 pacientes, por lo que es el segundo diagnóstico cardiológico más frecuente, y coincidiendo con miocardiopatía hipertrófica en 3 de ellos. Ambos siguen siendo diagnósticos que deben hacernos pensar en la posibilidad de que se encuentren en el contexto de una entidad malformativa más amplia. Igualmente, se sigue en consultas al paciente de 3 años en el que no se encontró cardiopatía congénita, para descartar la aparición evolutiva de anomalías cardiacas estructurales, como se ha descrito previamente33. Si bien en nuestra serie no se produjo ningún evento letal ni fue preciso colocar desfibriladores, se remitió al paciente de más edad a una unidad de cardiología para estudio por síncopes. De hecho, se ha descrito que los pacientes con SL con miocardiopatía hipertrófica parecen tener mayor riesgo de eventos adversos durante el seguimiento32. Entre las alteraciones cardiacas, si bien son frecuentes los defectos de conducción, el síndrome de Wolf-Parkinson-White descrito en uno de nuestros pacientes no es una manifestación común.
El solapamiento fenotípico entre el SL, el SN y la neurofibromatosis tipo 1 dificulta la correcta clasificación de estos pacientes. El síndrome de Noonan-neurofibromatosis, relacionado con mutaciones en el gen NF134 y caracterizado por la asociación de neurofibromatosis tipo 1 y manifestaciones del SN, presenta manifestaciones muy parecidas al SL. El diagnóstico de una cardiopatía congénita como la miocardiopatía hipertrófica en los pacientes con síndrome de Noonan-neurofibromatosis hace recomendable comenzar el estudio genético por PTPN11 en vez de por NF135. En las tres entidades son frecuentes la lentiginosis múltiple y las manchas café con leche. Así como las manchas café con leche aumentan en número y tamaño con la edad en estas entidades, la lentiginosis múltiple no es frecuente en los niños más pequeños. Por ese motivo, algunos autores recomiendan valorar el diagnóstico de SL cuando un paciente menor de 1 año con SN asocie manchas café con leche29. La distinción entre la lentiginosis y los nevos múltiples puede resultar particularmente compleja en algunos pacientes, y algunos autores proponen incluir las lesiones melanocíticas en el espectro del SL36. En nuestra serie se ha limitado el diagnóstico, cuando ha sido posible, a pacientes con lesiones lentiginosas. Así lo ilustra el caso de un paciente descrito en Ezquieta et al13. El paciente padecía SN por el cambio aminoacídico p.Asn308Asp en PTPN11 y tenía lesiones hiperpigmentadas múltiples, al igual que su madre, que era negativa para el estudio mutacional, lo que indica que se trataba de lesiones névicas múltiples presentes en ambos por una causa diferente de las mutaciones en PTPN11. Destaca que, como ya indicaban observaciones previas, hemos podido demostrar que el SL se asocia a una talla más alta que el resto de los SNCFC.
CONCLUSIONESLa estenosis pulmonar valvular y muy particularmente la miocardiopatía hipertrófica son hallazgos cardiológicos que pueden dar la pista fundamental para identificar el síndrome. Dadas las potenciales implicaciones clínicas de la miocardiopatía hipertrófica, se debe buscar activamente en los pacientes del espectro clínico del SN y muy especialmente en aquellos con SL. Los criterios originalmente descritos por Voron et al en 1976 siguen siendo de ayuda para la clasificación diagnóstica de estos pacientes; aunque a los portadores de anomalías asociadas al SL que no cumplan estos criterios, se les debe dar seguimiento evolutivo porque algunas de las manifestaciones cutáneas, en particular la lentiginosis múltiple, pueden tardar en manifestarse hasta bien entrada la pubertad.
FINANCIACIÓNFondo de Investigaciones Sanitarias (PI 06/1179).
CONFLICTO DE INTERESESNinguno.
Los autores agradecen la colaboración de los pacientes y sus familiares, así como a los clínicos que no han podido incluirse entre los autores, como los Dres. López-Siguero (Hospital Carlos Haya, Málaga), Barrio y García-Sagredo (Hospital Ramón y Cajal, Madrid), Kuburovic (Mother and Child Healthcare Institute, Serbia), Gracia (Hospital Universitario La Paz, Madrid), Gener Querol (Hospital Universitario de Cruces, Barakaldo, Vizcaya), González-Meneses (Hospital Virgen del Rocío, Sevilla) y Aleixandre Blanquer (Hospital General Universitario de Elda, Alicante). Begoña Ezquieta es investigadora adscrita de la U753 del CIBER de Enfermedades Raras.