
El síndrome de Shprintzen-Goldberg (SSG) es una afección del tejido conectivo que se debe incluir en el diagnóstico diferencial de síndromes aórticos como el de Marfan (SM) o Loeys-Dietz (SLD). El SSG tiene su causa en variantes patogénicas en el gen SKI, implicado en la vía de señalización del factor de crecimiento transformador beta (TGFβ)1,2. Hasta la fecha se han descrito menos de 100 pacientes con SSG confirmado. El fenotipo incluye dismorfismo craneofacial (como dolicocefalia/escafocefalia, frente prominente, proptosis, hipertelorismo, anomalías auriculares y microrretrognatia, entre otras), alteraciones esqueléticas, cutáneas y oculares, valvulopatías cardiacas, dilatación de la raíz aórtica, anomalías neurológicas, trastornos del comportamiento y déficit cognitivo en grado variable3–5.
Se presenta a 2 pacientes no relacionados y se discute su actitud diagnóstico-terapéutica con especial referencia a la indicación de cirugía aórtica.
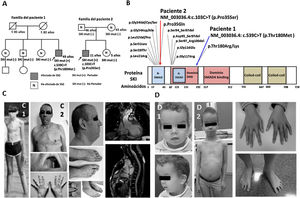
El primer caso se diagnosticó a la edad de 12 años de SM por criterios clínicos (puntuación sistémica ≥ 7). Padres no eran consanguíneos, y sus padres y su hermanos no presentaban el mismo fenotipo. A los 45 años se le detectó una dilatación de la raíz aórtica (45 mm) (figura 1) y se planteó el reemplazo quirúrgico profiláctico, dada la sospecha de SM. Se lo derivó a la unidad de cardiopatías familiares para estudio. Se realizó un estudio genético mediante secuenciación masiva (NGS) (35 genes, entre ellos FBN1), que identificó la variante p.Thr180Met en el gen SKI. En el paciente, además del hábito marfanoide, destacaba un dismorfismo craneofacial característico, sin discapacidad intelectual (tabla 1). Ninguno de sus hermanos era portador de la variante y, aunque no se estudió a los padres, fallecieron a edades avanzadas. Lo más probable es que se tratase de una variante de novo. Se diagnosticó SSG y se decidió una actitud expectante. Tras 4 años, no ha sufrido eventos y el diámetro aórtico continúa estable.
; resonancia magnética cardiovascular del paciente 1 (diámetro de la raíz aórtica, 45 mm). D: fenotipo del paciente 2 a los 4 y los 9 años (D1, D2); fotografías tomadas previo consentimiento informado. SSG: síndrome de Shprintzen-Goldberg.")
Estudio clínico-genético del probando y su familia. A: árbol genealógico de las familias y estudio genético. B: variantes descritas en el gen SKI a nivel de proteína. C: fenotipo del paciente 1 a los 12 y los 45 años (C1, C2); resonancia magnética cardiovascular del paciente 1 (diámetro de la raíz aórtica, 45 mm). D: fenotipo del paciente 2 a los 4 y los 9 años (D1, D2); fotografías tomadas previo consentimiento informado. SSG: síndrome de Shprintzen-Goldberg.
Fenotipos de los pacientes 1 y 2 y los portadores de variantes que afectan a los aminoácidos Thr180 y Pro35
| Paciente 1 | Portadores de variantes en AA p.Thr180a (n=9)5 | Paciente 2 | Portadores de variantes en AA p.Pro35b (n=6)2-4 | |
|---|---|---|---|---|
| Edad al diagnóstico | 45 años | Media, 15,8 (intervalo 2-47) años | 4 años | Media 17,5 (intervalo, 6-46) años |
| Edad al último seguimiento | 49 años | Media, 21,8 (intervalo, 3-49) años | 11 años | ND |
| Variantes en el gen SKI | c.539C> T, p.Thr180Met | p.Thr180Met (5), p.Thr180Lys (3), p.Thr180Arg (1) | c.103C> T, p.Pro35Ser | p.Pro35Ser (5),p.Pro35Gln (1) |
| Herencia | Esporádico (sin muestras de los padres) | 7 de novo2 esporádicos (sin muestras parentales) | De novo | 6 de novo |
| Craniosinostosis | Braquicefalia | 0/7, 2 ND | Escafocefalia | 6/6 |
| Proptosis ocular | Sí | 4/8, 1 ND | Sí | 6/6 |
| Hipertelorismo | Sí | 6/8, 1 ND | Sí | 6/6 |
| Anomalías de agudeza visual | No | 9 ND | Miopía y astigmatismo | 1/6 |
| Paladar ojival | Sí | 8/8, 1 ND | Sí | 4/4, 2 ND |
| Anomalías de las orejas | Pabellones auriculares grandes | 1/7, 2 ND | Orejas levantadas | 1/1, 5 ND |
| Hipoplasia malar | Si | 5/8, 1 ND | Sí | 6/6 |
| Micrognatia o retrognatia | Si | 5/9 | Sí | 6/6 |
| Prolapso/insuficiencia valvular mitral | No | 4/9 | Insuficiencia valvular mitral | 3/6 |
| Dilatación aórtica | Dilatación de raíz aórtica (45 mm) | 6/9 | No (raíz aórtica de 24 mm) | 1/6 |
| Tortuosidad arterial | No | 0/4, 5 ND | No | 1/6 (portador de p.Pro35Gln): carótida interna y vertebrobasilar |
| Deformidad torácica | Pectus carinatum | 8/9 | Pectus excavatum | 3/3, 3 ND |
| Aracnodactilia | Sí | 8/9 | Sí | 6/6 |
| Talla alta (para la edad) | Sí | ND | Sí | ND |
| Dolicostenomelia | Sí | 6/9 | Sí | 6 ND |
| Osteopenia | Sí | 9 ND | No | 6 ND |
| Escoliosis o cifosis | Cifoescolisosis grave | 4/8, 1 ND | No | 4/5, 1 ND |
| Laxitud articular | Si | 8/9 | No | 3/3, 3 ND |
| Contracturas articulares | No | 4/8, 1 ND | Sí, camptodactilia | 5/5, 1 ND |
| Deformidad de pies | Sí (bilateral corregida) | 9/9 | Sí | 6 ND |
| Protrusión acetabular | No | 4/5, 4 ND | Báscula pélvica por dismetría de fémur derecho | 6 ND |
| Pies planos | Sí | 9/9 | No | 6 ND |
| Anomalías cutáneas | Estrías, piel grasa con acné comedónico | 3/9 | Piel traslúcida | 3/5 hernia inguinal,1 ND |
| Anomalías venosas | Venas varices en EI | 9 ND | No | 6 ND |
| Hipotonía | Sí | 2/9 | No | 1/1, 5 ND |
| Otras anomalías neuromusculares | No | 9 ND | Torpeza psicomotriz | No |
| Anomalías cerebrales | ND | 9 ND | Displasia de cuerpo calloso e hipocampo | 6 ND |
| Ectasia dural | No | 9 ND | No | 1/1, 5 ND |
| Déficit cognitivo | No | 0/8, 1 ND | Leve | 6/6 |
| Problemas de aprendizaje | No | 3/8, 1 ND | Sí | 6 ND |
| Trastorno del lenguaje | Si | 9 ND | Sí | 6 ND |
| Trastornos del comportamiento | ND | 9 ND | TDAH | 6 ND |
| Cáncer | No | 9 ND | Hepatoblastoma | 6 ND |
| Otras anomalías | No | ND | Hipertrofia del hemicuerpo derecho | ND |
EI: extremidades inferiores; ND: no determinado; TDAH: trastorno de déficit de atención e hiperactividad.
La variante p.Thr180Met se ha publicado previamente en 5 casos esporádicos de SSG, clasificada como patogénica5. Se han descrito otras 2 variantes en 4 pacientes con SSG que afectan al mismo aminoácido. En total se describe a 9 portadores afectados, de los que 6 presentaron dilatación aórtica entre los 15 a 47 años: se trató con cirugía solo a 1 de ellos a los 47 años, por alcanzar un diámetro de 59mm. El déficit cognitivo parece ser leve o ausente en estos portadores.
El segundo caso se derivó a genética a los 4 meses de edad por dismorfia craneofacial y ventriculomegalia de diagnóstico prenatal. Se evidenció hernia umbilical e hemihipertrofia corporal y por ecografía se identificó hepatoblastoma. Se descartaron síndromes de Beckwith-Wiedemann y Otopalatodigital molecularmente. A los 4 años se observaban hábito marfanoide, alteraciones esqueléticas y trastorno de aprendizaje, y se sospechó SSG (figura 1). Se realizó estudio genético mediante secuenciación Sanger del gen SKI, que identificó la variante p.Pro35Ser. Actualmente, con 11 años, no se ha detectado dilatación aórtica (raíz aórtica de 24 mm).
La variante p.Pro35Ser se ha publicado como patogénica en 5 casos esporádicos de SSG. Se ha descrito otra variante en 1 paciente con SSG (tabla 1) que afecta al mismo aminoácido. En total se ha descrito a 6 portadores afectados de entre 6 y 46 años, y solo el portador de la otra variante (p.Pro35Gln) requirió intervención quirúrgica por dilatación de la raíz aórtica con 16 años (Z-score=7,01, no especifican el diámetro aórtico)2–4.
El SSG presenta un fenotipo complejo que implica el diagnóstico diferencial con SM y SLD. En estos síndromes, la indicación quirúrgica de la dilatación aórtica está claramente establecida. Sin embargo, en el SSG no existen recomendaciones claras. Hasta la fecha, en el SSG no se han descrito casos de muerte con disección aórtica. Sin presencia de factores de riesgo, se recomienda el seguimiento estrecho de los diámetros aórticos. La cirugía profiláctica se podría indicar según algunas recomendaciones de expertos (en la franja de los 50-55mm o con una tasa de progresión> 5 mm/año6). En ausencia de estudios amplios en esta enfermedad de muy baja prevalencia, será necesario disponer de más casos clínicos descritos.
La implementación clínica de paneles de genes mediante NGS en el estudio de las aortopatías sindrómicas facilita el análisis de estas enfermedades solapadas que implican eventos cardiovasculares adversos, que van desde menos graves a muy graves o múltiples y requieren medidas clínico-quirúrgicas más agresivas.
CONFLICTO DE INTERESESL. Monserrat es accionista de la Empresa de Genética Health in Code. Los demás autores manifiestan no tener conflictos de intereses.
Especialmente a los pacientes y a sus familias por compartir su información clínica e imágenes para este artículo. A la Dra. Carme Vila Obradors (Equip d’assistència primària EAP de Sant Quirze del Vallès) por su compromiso desde la pediatría con las enfermedades raras.