Sra. Editora:
El síndrome de QT largo se caracteriza por una prolongación del intervalo QT en el electrocardiograma (ECG), definido como un QT corregido > 450 ms en varones y > 470 ms en mujeres1, que predispone al desarrollo de arritmias ventriculares del tipo torsade de pointes. Se diferencian dos grandes grupos: el QT largo congénito, asociado con mutaciones en determinados genes, y la variante adquirida, asociada con factores ambientales2.
La principal causa de QT largo adquirido es farmacológica, y hay gran variedad de fármacos asociados con prolongación del intervalo QT3. Otras causas incluyen los trastornos electrolíticos (fundamentalmente hipopotasemia, hipomagnesemia e hipocalcemia), tóxicos como los organofosforados, dietas proteicas líquidas, trastornos endocrinos como el hipotiroidismo4 o el feocromocitoma, la inanición, la anorexia nerviosa o en relación con bradiarritmias.
Presentamos el caso de un varón de raza blanca y 50 años de edad que ingresó por haber sufrido un episodio sincopal mientras conducía, con accidente de tráfico secundario a aquel. Como único antecedente de interés presentaba hipertensión arterial diagnosticada 3 meses antes, por lo que había iniciado tratamiento con amlodipino 5, valsartán 160 e hidroclorotiazida 12,5, con adecuado control. Desde entonces refería sensación continua de sed, poliuria y polidipsia, sin haber presentado previamente mareo ni síncope.
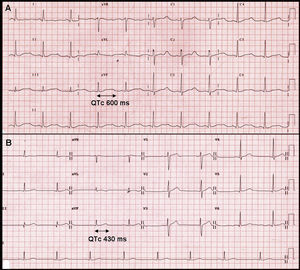
A su llegada a urgencias, se encontraba consciente y orientado, con Glasgow 15, presión arterial de 150/90mmHg y frecuencia cardiaca en 59 lpm. La exploración cardiorrespiratoria y neurológica fue normal. En el ECG se objetivó ritmo sinusal con una prolongación muy significativa del intervalo QT, y el QT corregido mediante la fórmula de Bazzet de 600ms (Figura 1A). En la analítica destacaba una marcada alteración electrolítica con hipopotasemia (2,1 mEq/l) e hipernatremia (150 mEq/l). El magnesio era normal y la gasometría arterial mostraba ligera alcalosis (pH 7,5). Ante estos hallazgos, se ingresó al paciente en la unidad de intermedios cardiológicos para monitorización con la sospecha inicial de trastorno del eje renina-angiotensina-aldosterona como causa del cuadro.

Figura 1. A: electrocardiograma en presencia de intensa hipopotasemia con prolongación del intervalo QT. B: electrocardiograma tras la resolución de la hipopotasemia con intervalo QT normal.
Se necesitaron altas dosis de suplementos de potasio para corregir la hipopotasemia, con normalización progresiva de la duración del intervalo QT hasta recuperar valores dentro de la normalidad (Figura 1B). Para el control de la hipertensión arterial se utilizaron inicialmente antagonistas del calcio y bloqueadores alfa, que obtuvieron un control subóptimo, y fue necesario añadir inhibidores de la enzima de conversión de la angiotensina y antialdosterónicos, una vez realizados los estudios hormonales, para lograr un adecuado control.
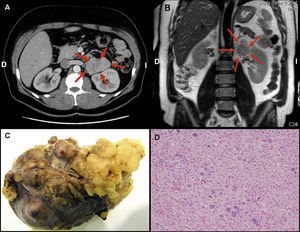
Se realizó un ecocardiograma transtorácico, que descartó una cardiopatía estructural. Con vistas al diagnóstico etiológico, se realizó un estudio de función suprarrenal —que mostró concentraciones plasmáticas de aldosterona elevadas, con actividad de la renina plasmática suprimida— y una tomografía computarizada toracoabdominal, que evidenció una tumoración homogénea lobulada de 6,8 × 6,2 × 4 cm dependiente de la glándula suprarrenal izquierda (Figura 2A) y que no afectaba a otros órganos, por lo que se llegó al diagnóstico de hiperaldosteronismo primario en relación con masa suprarrenal izquierda.

Figura 2. Imagen de tomografía computarizada (A) y resonancia magnética (B) de la masa suprarrenal izquierda. Imagen macroscópica (C) y microscópica (D) de la tumoración. D: derecha; I: izquierda.
En la monitorización presentó inicialmente abundantes extrasístoles ventriculares de diferentes morfologías, con dobletes y tripletes, pero sin eventos arrítmicos sostenidos, que fueron remitiendo progresivamente con la normalización del QT. En situación estable, fue dado de alta con suplementos de potasio y el tratamiento antihipertensivo, pendiente de intervención quirúrgica de la masa suprarrenal.
El paciente se mantuvo asintomático, y se realizó un control analítico estricto para mantener los iones dentro de la normalidad. Se realizó una resonancia magnética preoperatoria, que demostró un realce heterogéneo de la tumoración con gadolinio y la presencia de pequeñas adenopatías en retroperitoneo (Figura 2B). De manera programada, se intervino quirúrgicamente al paciente para exéresis de la tumoración suprarrenal izquierda y linfadenectomía asociada. La anatomía patológica (Figura 2C y D) caracterizó la masa como carcinoma de corteza suprarrenal sin evidencia de metástasis en los ganglios linfáticos. Al microscopio, presentaba una arquitectura variable; estaba formado por células de citoplasma claro con importante pleomorfismo nuclear, zonas de necrosis y mitosis frecuentes, algunas atípicas. Además se observó invasión de la cápsula, sin invasión venosa. Tras la intervención, se normalizaron los electrolitos y las cifras de presión arterial, por lo que se pudo suspender tanto los suplementos de potasio como el tratamiento antihipertensivo.
Nuestro paciente presentaba una prolongación adquirida del intervalo QT en relación con una intensa hipopotasemia, y recuperó los valores normales tras corregirse el trastorno electrolítico. En este contexto clínico, asumimos que el síncope del paciente fue secundario a un evento arrítmico del tipo torsade de pointes en relación con la intensa hipopotasemia. Como etiología de esta, se confirmó la sospecha inicial y se llegó al diagnóstico de hiperaldosteronismo primario secundario a carcinoma de corteza suprarrenal.
El carcinoma suprarrenal es una entidad muy agresiva y poco frecuente, con una incidencia descrita de 1-2 casos/millón de habitantes/año5. Aproximadamente un 60% de ellos son funcionantes, y la presentación más frecuente es el síndrome de Cushing. El carcinoma suprarrenal como causa de hiperaldosteronismo primario es extremadamente raro, con una incidencia de un 1-3% del total de casos6. La resección quirúrgica completa tanto del tumor como de las adenopatías locales es el único tratamiento que se ha demostrado eficaz. La presentación como hiperaldosteronismo primario podría determinar un diagnóstico más temprano y permitir una intervención precoz sobre la neoplasia.
Autor para correspondencia: lgarbue@googlemail.com