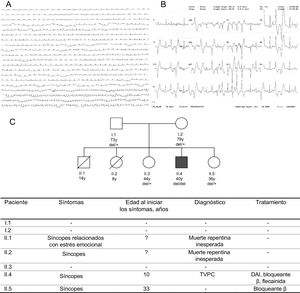
Se presenta el caso de un hombre de 40 años con antecedentes de síncope desde la infancia. Desde los 10 años ha sido tratado con 2,5mg de carteolol dos veces al día, a causa del síncope y la arritmia ventricular. Cuando tenía 17 años, se le cambió el bloqueante β y empezó con nadolol, 80mg cada día, que el paciente dejó de tomar voluntariamente. Ingresó en el hospital tras sufrir un síncope mientras caminaba. Se constató la existencia de antecedentes familiares de muerte súbita cardiaca: un hermano y una hermana fallecieron repentinamente a la edad de 8 y 14 años, respectivamente. Los padres aparentemente estaban sanos y los abuelos murieron a edad avanzada a causa de un tumor. Los resultados de las pruebas analíticas, el electrocardiograma, el intervalo QT, la exploración ecocardiográfica y la cardiorresonancia magnética eran normales. En los registros del electrocardiograma se detectó una taquicardia ventricular polimórfica (TVP) intermitente. La prueba de esfuerzo sobre cinta rodante reveló TVP intermitente durante el esfuerzo máximo que desaparecía en reposo (figura 1A y figura 1B). Se estableció un diagnóstico de taquicardia ventricular polimórfica catecolaminérgica (TVPC) debida a TVP inducida por el ejercicio en presencia de un corazón estructuralmente normal y un electrocardiograma normal. Se restableció el tratamiento con bloqueantes β y se repitió la prueba de esfuerzo sobre cinta rodante. La persistencia de la TVP intermitente durante el ejercicio hizo que se le implantara un desfibrilador automático implantable con retrasos prolongados antes de la descarga cardiaca. Durante el seguimiento, se añadió flecainida, a una dosis de 100mg dos veces al día, al tratamiento con bloqueantes β debido a las descargas apropiadas. Tras un seguimiento de 6 meses, el control de la arritmia ventricular mejoró notablemente sin episodios sostenidos.
. B: ECG de 12 derivaciones durante una prueba de esfuerzo sobre cinta rodante con ataques de TVP. C: esquema genealógico familiar. Se indica genotipo para la deleción CASQ2. DAI, desfibrilador automático implantable; ECG, electrocardiograma; TVPC, taquicardia ventricular polimórfica catecalominérgica.")
A: registro telemetría ECG que muestra ectopia ventricular muy frecuente (abajo). B: ECG de 12 derivaciones durante una prueba de esfuerzo sobre cinta rodante con ataques de TVP. C: esquema genealógico familiar. Se indica genotipo para la deleción CASQ2. DAI, desfibrilador automático implantable; ECG, electrocardiograma; TVPC, taquicardia ventricular polimórfica catecalominérgica.
Se realizó una prueba genética para confirmar el diagnóstico1–3. Se recogieron muestras de sangre periférica del caso índice y de cuatro familiares (figura 1C). Lamentablemente, no era posible disponer de muestras biológicas de los familiares fallecidos. En el caso índice4 se realizó la secuenciación masiva de los 28 genes asociados con enfermedad arritmogénica.
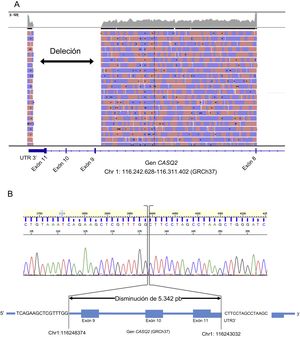
La secuenciación mostró que no había variante causal de nucleótido simple pero constató la ausencia de la secuencia para los tres últimos exones del gen CASQ2. Se llevó a cabo la reacción en cadena de la polimerasa de gran alcance para amplificar un fragmento de aproximadamente 18kb, donde se localizaba la deleción. Los resultados confirmaron la presencia de un fragmento de aproximadamente 13kb en el caso índice y un fragmento de aproximadamente 18kb en un muestra de control. El producto de la reacción en cadena de la polimerasa del caso índice se secuenció con el sistema Ion Proton (Thermo Fisher Scientific). Los resultados demostraron la presencia de una deleción de aproximadamente 5kb causante de la pérdida de los exones 9, 10 y 11 del gen CASQ2 (figura 2). La localización exacta de la deleción se confirmó utilizando una reacción en cadena de la polimerasa específica y mediante secuenciación Sanger (NM_001232:c.839-461_*830del).
 para el producto de la RCP de gran alcance del caso índice. B: electroferograma y representación de la localización exacta de la deleción.")
El caso índice de la familia en cuestión mostró una deleción homocigótica clasificada como patógena según el American College of Medical Genetics (ACMG)5, que confirmaba el diagnóstico de TVPC, mientras que los familiares no afectados eran portadores heterocigóticos de la deleción (figura 1C). Una de las hermanas había experimentado hacía poco síncope de causa desconocida. Basándose en la hipótesis de un mayor riesgo de arritmias asociado con las variantes heterocigóticas, se la trató con bloqueantes β.
La TVP catecolaminérgica es un trastorno arritmogénico hereditario que se caracteriza por taquicardia ventricular bidireccional inducida por adrenérgicos o TVP. Se han identificado dos tipos genéticos: una variante dominante debida a mutaciones en el gen del receptor cardiaco de rianodina (RyR2) y una variante recesiva poco frecuente causada por mutaciones en el gen de la calsequestrina (CASQ2)1,3. Se han identificado mutaciones en otros genes tales como KCNJ2, ANK2, TRDN y CALM1 en pacientes con características clínicas parecidas a la TVPC pero no está claro si estas son fenocopias de la TVPC3. La mayor parte de las mutaciones descritas en el gen CASQ2 son variantes genéticas de corte y empalme y, hasta la fecha, no existen informes de variaciones causales en el número de copias. Aunque se han descrito algunas variaciones en el número de copias en las canalopatías, no son habituales; en el caso concreto de la TVPC, se ha descrito una deleción del exón 3 de RyR2.
Las manifestaciones clínicas tienen lugar en la primera década de la vida y son desencadenadas por la actividad física o el estrés emocional. El tratamiento de primera línea consiste en limitar el ejercicio y administrar bloqueantes β sin actividad simpaticomimética. Datos preliminares sugieren que la flecainida reduce la carga de arritmia ventricular en algunos pacientes y debería considerarse su adición a los bloqueantes β3. El diagnóstico se confirma mediante pruebas genéticas. Las tecnologías de secuenciación masiva permiten el cribado rápido del nucleótido simple o de pequeñas variantes “indel” (de inserción o deleción). No obstante, la presencia de deleciones o inserciones grandes puede pasar desapercibida, si solo se tienen en cuenta las listas de variantes descritas por los programas de búsqueda de variantes. La presencia de estas inserciones o deleciones debe comprobarse con programas específicos, de lo contrario los resultados de la secuenciación deberían revisarse con visores genómicos, comprobando la cobertura de todas las regiones objetivo para la secuenciación4.
Este es el primer caso comunicado de una variación en el número de copias como causa de la TVPC en una familia sin consanguinidad con mal pronóstico, con un caso índice gravemente afectado portador de la variante en homocigosis y con dos hermanos fallecidos repentinamente a edades muy tempranas.
FINANCIACIÓNEste estudio contó con el apoyo del Plan Estatal de I+D+i 2008-2011 y 2013-2016, Subdirección General de Evaluación y Fomento de la Investigación (ISCIII-SGEFI) del Instituto de Salud Carlos III (ISCIII) y del Fondo Europeo de Desarrollo Regional (FEDER) (subvenciones número PI16/00903, RD12/0042/0037, CB16/11/00226).