La taquicardia ventricular polimórfica catecolaminérgica (TVPC) es una cardiopatía hereditaria caracterizada por la aparición de taquicardia ventricular polimórfica durante el esfuerzo, las emociones o la perfusión de catecolaminas1. A esta cardiopatía se la considera una enfermedad rara, con una prevalencia de 1/10.000 habitantes, y es una entidad altamente letal (el 30% de muertes súbitas en menores de 40 años sin bloqueadores beta)2. Suele ser de herencia autosómica dominante, con penetrancia del 80%, y las mutaciones frecuentemente afectan al gen del receptor de la rianodina (RyR2)3. Para el diagnóstico suelen ser necesarios la prueba de esfuerzo (PE) o la de adrenalina4–6.
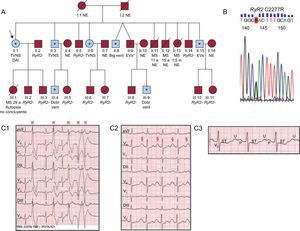
Nuestro objetivo es describir una familia de 19 miembros vivos (figura A), con 4 muertes súbitas, 8 portadores de la mutación nueva RyR2 C2277R (genotipo+) (figura B) y 7 de ellos con el fenotipo de TVPC desenmascarado por la PE (fenotipo+).
 del primer minuto de la recuperación de la prueba de esfuerzo de la probando, que demuestra arritmias ventriculares polimórficas (C1, ) y fusión de onda U marcada con la onda P posterior (C2, §). a: años; Big vent: bigeminismo ventricular; DAI: desfibrilador automático implantable; Dobl vent: dobletes ventriculares; EVs: extrasístoles ventriculares; MS: muerte súbita; NE: no evaluado; m: meses; TVNS: taquicardia ventricular no sostenida. Esta figura se muestra a todo color solo en la versión electrónica.")
A: árbol familiar; la flecha apunta a la probando; los símbolos azules representan a pacientes con fenotipo+ de taquicardia ventricular polimórfica catecolaminérgica y los asteriscos destacan el estado de portador heterocigoto de la mutación RyR2 C2277R. B: electroferograma del fragmento del exon 45 del gen RyR2 que contiene la mutación. C: registros consecutivos (C1-C3) del primer minuto de la recuperación de la prueba de esfuerzo de la probando, que demuestra arritmias ventriculares polimórficas (C1,
La probando (II:1), de 56 años, consultó por síncopes y palpitaciones. Refería la muerte súbita de tres hermanos con 11 y 15 años (por ejercicio físico y una discusión) y con 1,5 meses de vida, así como de una hija de 29 años mientras bailaba (con síncopes previos de esfuerzo), aunque solo a esta se le practicó autopsia, que resultó no concluyente. Aplicamos el protocolo de estudio familiar tras muerte súbita sin causa aclarada aprobado por nuestro comité ético, y cada individuo firmó el consentimiento informado. En la probando, el electrocardiograma y el ecocardiograma resultaron normales. Su PE (protocolo de Bruce) mostró arritmias ventriculares a partir de 100 lpm y fue diagnóstica de TVPC (figura C). Este diagnóstico se definió por la presencia de dobletes ventriculares, taquicardias ventriculares sostenidas o taquicardias ventriculares no sostenidas polimórficas o > 10 extrasístoles ventriculares/min durante la PE o el test de adrenalina6. Otros 6 miembros, de los 19 evaluados, presentaron una respuesta similar, con diferentes grados de complejidad de las arritmias ventriculares (figura A, tabla). Ante la sospecha de TVPC autosómica dominante, secuenciamos los 33 exones más frecuentemente afectados en el gen de RyR2 en la probando. Identificamos una variante missense en heterocigosis en el exón 45 (C2277R), no descrita previamente en la literatura, localizada en un hot spot que codifica parte del dominio de unión a la proteína calstabina, que se catalogó como mutación probablemente asociada con la enfermedad. La mutación cosegregó con el fenotipo de TVPC, aunque en un caso de genotipo+ (II:9), en quien también realizamos un test de adrenalina (protocolo de la Clínica Mayo), las arritmias ventriculares no alcanzaron el criterio diagnóstico. Así, obtuvimos una cohorte de 8 sujetos con genotipo+, 7 de ellos con fenotipo+, desenmascarado durante la PE (penetrancia del 87,5%). El 75% de los sujetos con genotipo+ eran varones, con media de edad de 46 ± 10 años; el 37% refería síntomas arrítmicos previos y el 100% presentaba electrocardiograma normal en reposo (frecuencia cardiaca, 63 ± 10 lpm; QTc, 400 ± 27 ms). En la PE inicial, la frecuencia cardiaca máxima fue de 150 ± 15 lpm y la duración, 9 ± 2 min, y el diagnóstico se estableció a una frecuencia cardiaca de 132 ± 11 lpm.
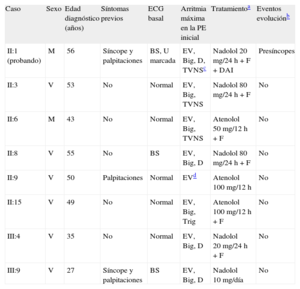
Características clínicas de la cohorte de ocho familiares portadores de la mutación en RyR2
| Caso | Sexo | Edad diagnóstico (años) | Síntomas previos | ECG basal | Arritmia máxima en la PE inicial | Tratamientoa | Eventos evoluciónb |
| II:1 (probando) | M | 56 | Síncope y palpitaciones | BS, U marcada | EV, Big, D, TVNSc | Nadolol 20 mg/24 h + F + DAI | Presíncopes |
| II:3 | V | 53 | No | Normal | EV, Big, TVNS | Nadolol 80 mg/24 h + F | No |
| II:6 | M | 43 | No | Normal | EV, Big, TVNS | Atenolol 50 mg/12 h + F | No |
| II:8 | V | 55 | No | BS | EV, Big, D | Nadolol 80 mg/24 h + F | No |
| II:9 | V | 50 | Palpitaciones | Normal | EVd | Atenolol 100 mg/12 h | No |
| II:15 | V | 49 | No | Normal | EV, Big, Trig | Atenolol 100 mg/12 h + F | No |
| III:4 | V | 35 | No | Normal | EV, Big, D | Nadolol 20 mg/24 h + F | No |
| III:9 | V | 27 | Síncope y palpitaciones | BS | EV, Big, D | Nadolol 10 mg/día | No |
Big: bigeminismo; BS: bradicardia sinusal; D: dobletes; DAI: desfibrilador automático implantable; ECG: electrocardiograma; EV: extrasistolia ventricular; F: flecainida; M: mujer; PE: prueba de esfuerzo; Trig: trigeminismo; TVNS: taquicardia ventricular no sostenida con cambio de eje de 180°; V: varón.
Los sujetos con genotipo+ fueron tratados con bloqueadores beta a la dosis máxima tolerada y, aunque en 3 (37%) desaparecieron las arritmias ventriculares en la PE evolutiva, en 5 (63%) persistió una carga arrítmica suficiente (extrasístoles ventriculares frecuentes, bigeminismo, dobletes, taquicardia ventricular no sostenida) para agregar flecainida, tal y como se ha propuesto en la literatura1 (tabla). En la probando se implantó un desfibrilador porque presentó presíncopes con taquicardia ventricular no sostenida en la PE a pesar del tratamiento con bloqueadores beta máximo (antes del inicio del uso de flecainida en este contexto clínico1). Finalmente, a los 34 ± 4 meses de seguimiento, todos los pacientes estaban asintomáticos, sin carga arrítmica ni eventos clínicos destacables (muerte súbita, síncopes o descarga apropiada del desfibrilador).
En definitiva, describimos por primera vez la mutación RyR2 C2277R, causante de TVPC, en una familia con una alta letalidad en jóvenes, con un buen rendimiento diagnóstico de la PE y una excelente respuesta al tratamiento con bloqueadores beta con y sin flecainida.
FINANCIACIÓNEste trabajo ha sido financiado por el Instituto de Salud Carlos III (PI14/01477 y RD12/0042/0029), Prometeo 2011/027, Sociedad Española de Cardiología (beca Pedro Zarco) y Agence Nationale de la Recherche (ANR-13-BSV1-0023-03).
Agradecemos la amable colaboración de los pacientes y del grupo del trabajo de muerte súbita infantil de la Asociación Española de Pediatría, así como el soporte técnico del Biobanco La Fe (PT13/0010/0026).