La enfermedad de Fabry (EF) es un trastorno del almacenamiento lisosómico progresivo y poco frecuente, causado por una deficiencia funcional de la enzima alfagalactosidasa (α-gal A) lisosómica1. Se trata de un trastorno hereditario ligado al cromosoma X con origen en variantes genéticas patógenas presentes en el gen GLA. Dichas variantes producen una deficiencia enzimática funcional que resulta en una acumulación intracelular de glucoesfingolípidos, predominantemente globotriaosilceramida (liso-Gb3) en los compartimentos lisosómicos y no lisosómicos de las células de la piel, el corazón, el cerebro y otros tejidos, lo cual contribuye a producir la manifestación multisistémica de este trastorno y la muerte temprana del paciente1,2.
La enfermedad de Fabry se describió por primera vez en pacientes varones con una forma grave de la enfermedad, un fenotipo que actualmente se denomina forma clásica de la enfermedad1,2. Estos pacientes suelen ser portadores de variantes genéticas con cambio de sentido o con desplazamiento del marco de lectura que generan ausencia o reducción grave de la actividad de α-gal A, con inicio de los síntomas durante la infancia o la adolescencia, insuficiencia progresiva de múltiples órganos y, finalmente, muerte temprana1–3. Sin embargo, un grupo mucho más amplio de pacientes suelen ser portadores de otras variantes genéticas que no causan estas alteraciones tan importantes en la estructura proteica (como ocurre con la mayoría de las mutaciones de cambio de sentido) y dan lugar a un grado variable de actividad residual de α-gal A, lo cual podría explicar los fenotipos de inicio tardío y menos graves observados en la expresión fenotípica del trastorno3.
Las mujeres son heterocigotas para las variantes genéticas de GLA y manifiestan un espectro clínico heterogéneo que va de la ausencia de síntomas a una gravedad similar a la de los pacientes varones. La gravedad depende en parte de la variante genética y el patrón de inactivación del cromosoma X, lo que se denomina lionización y es un proceso aleatorio. En consecuencia, las pacientes heterocigotas que expresan predominantemente un alelo de GLA no mutado sufren pocos síntomas o ninguno, mientras que las pacientes que expresan predominantemente un alelo de GLA mutado pueden presentar una progresión del trastorno similar a la de los fenotipos masculinos, ya sea en una forma clásica o en otra de inicio tardío, dependiendo de la variante genética subyacente en la familia4. A través de esta inactivación aleatoria del alelo mutado, en las mujeres la actividad de la alfagalactosidasa y la concentración de liso-Gb3 podrían presentar valores normales.
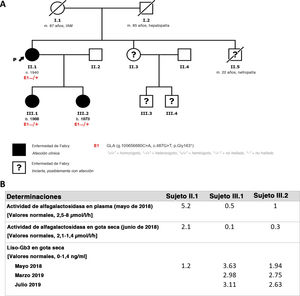
Se presenta el caso de una mujer de 78 años que estaba en seguimiento a causa de una miocardiopatía hipertrófica (MCH), con múltiples episodios de arritmias supraventriculares y ventriculares y portadora de un desfibrilador automático implantable en prevención primaria. Cursaba también con enfermedad renal crónica en estadio 4A2, con múltiples quistes renales y proteinuria, además de depósitos corneales atribuidos al tratamiento crónico con amiodarona. Para completar el estudio, se obtuvo un panel genético de MCH (ultrasecuenciación, 104 genes) que detectó una variante genética patógena del gen GLA (p.Gly163*), lo cual permitió establecer un diagnóstico de EF con afección cardiaca, renal y ocular (cornea verticillata). Se determinó la actividad enzimática y los liso-Gb3, cuyos valores estaban dentro de los límites normales (figura 1B).

En el estudio familiar (figura 1A), se identificó a un hermano que había fallecido a los 22 años a causa de un trastorno renal y a una hermana y 2 sobrinos con trastornos psiquiátricos que no quisieron participar en el estudio. Se evaluó a las 2 hijas de la paciente índice, de 52 y 47 años, que son portadoras de la misma variante genética y tienen EF. Ninguna de estas 2 pacientes tiene hijos. La hija de mayor edad sufre una afección renal en estadio 2A2 con proteinuria significativa e hipertrofia ventricular izquierda (figura 2B,C). La hija de menor edad presenta una afección renal leve con microalbuminuria y una ligera afección ocular. A la exploración física, no se observaron lesiones cutáneas o síntomas de acroparestesia. En ambas pacientes la actividad enzimática estaba reducida y los análisis mostraron también altas concentraciones de liso-Gb3 en ambas (figura 1B). En la biopsia renal, se observó una acumulación lisosomal de lípidos compatible con los cuerpos de cebra característicos de la EF (figura 2D). Además, no se observaron anomalías en los exámenes neurológicos y otorrinolaringológicos. Se inició un tratamiento de sustitución enzimática para las 2 pacientes jóvenes, sin complicaciones ni efectos adversos y sin que se observara progresión de las afecciones renal o cardiaca en un seguimiento de 2 años.
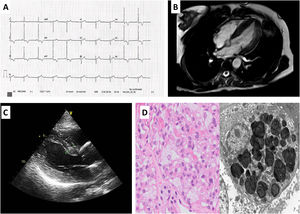
 de glomérulo con tinción de hematoxilina y eosina; podocitos agrandados a causa de la vacuolización citoplasmática. Microscopia electrónica (derecha) que muestra abundantes inclusiones citoplasmáticas laminadas (figuras de mielina o cuerpos de cebra).")
A: electrocardiograma de la hija mayor de nuestro caso índice, en el que se observa un intervalo PR corto, hipertrofia del ventrículo izquierdo e isquemia subepicárdica. B y C: resonancia magnética cardiaca y ecocardiografía con hipertrofia del ventrículo izquierdo. D: microscopia óptica (izquierda) de glomérulo con tinción de hematoxilina y eosina; podocitos agrandados a causa de la vacuolización citoplasmática. Microscopia electrónica (derecha) que muestra abundantes inclusiones citoplasmáticas laminadas (figuras de mielina o cuerpos de cebra).
La variante genética hallada en esta familia (p.Gly163*) no se ha descrito anteriormente, pero es una mutación con cambio de sentido, que por lo general son variantes genéticas patógenas, ya que producen un codón de parada prematura que da lugar a una transcripción aberrante o péptidos truncados. Se dispone de información clínica sobre al menos 67 variantes genéticas del mismo tipo claramente ligadas a la EF. De ellas, se han incluido las que son similares a nuestro caso en un estudio de mujeres diagnosticadas de EF5 en el que se observó que casi todas estaban sintomáticas y presentaban signos de enfermedad multisistémica.
El diagnóstico de EF constituye un verdadero reto y a menudo es el cardiólogo quien la sospecha en casos de MCH. La EF es una enfermedad con múltiples manifestaciones que a menudo retrasan el diagnóstico y requiere un abordaje multidisciplinario que permita identificar pronto a esos pacientes y establecer la mejor forma de tratarlos. En el diagnóstico diferencial entre la MCH y la EF, son necesarios un alto grado de sospecha clínica y técnicas de imagen, además de la utilidad del estudio genético. Nuestro caso ilustra cómo un estudio genético con ultrasecuenciación es crucial para el diagnóstico de los casos de pacientes que presentan un inicio tardío o poca afección cardiaca.
Los autores confirman que se ha obtenido el consentimiento por escrito de los pacientes para la presentación y publicación de este artículo, incluidas las imágenes y sus textos correspondientes. También ha sido autorizado por el comité de ética.
FINANCIACIÓNNo se dispuso de ninguna financiación en relación con este trabajo.
CONTRIBUCIÓN DE LOS AUTORESA. Robles-Mezcua ha obtenido y analizado los datos y ha elaborado la versión inicial del documento. L. Morcillo-Hidalgo y M. Martín-Velázquez han participado en la obtención y el análisis de los datos. M. León-Fradejas ha llevado a cabo el estudio anatomopatológico de las muestras de biopsia y el informe correspondiente. J.M. García-Pinilla ha revisado los datos obtenidos y su análisis y ha corregido la versión final del documento.
CONFLICTO DE INTERESESNo se declara ninguno.