Se han descrito previamente variantes genéticas del gen HCN4 con el fenotipo combinado de síndrome del seno enfermo (SSE) y miocardiopatía no compactada del ventrículo izquierdo (MCNC). Actualmente hay pocos casos en los que se haya probado esta relación y ningún caso previo se ha relacionado con dilatación de la aurícula izquierda (DAi). El objetivo es estudiar un trastorno familiar caracterizado por SSE, DAi e hipertrabeculación/fenotipo de MCNC para identificar las bases genéticas y electrofisiológicas subyacentes.
MétodosSe realizó una evaluación clínica, genética y electrofisiológica de una familia con SSE y MCNC. Se practicaron electrocardiograma, Holter, ecocardiografía y ergometría, así como resonancia magnética en los casos patológicos. Se realizaron pruebas genéticas con técnicas next-generation-sequencing (NGS) y un estudio funcional de la variante genética candidata en células de ovario de hámster chino.
ResultadosSe observó bradicardia sinusal en 12 familiares, asociada con criterios diagnósticos de MCNC en 4, hipertrabeculación en 6 y DAi en 9. Se detectó la variante genética HCN4 c.1123C> T (p.Arg375Cys) en heterocigosis en todos los pacientes afectados y ausente en individuos normales. Los análisis electrofisiológicos mostraron que la amplitud y las densidades de las corrientes de HCN4 (IHCN4) generadas por canales HCN4 con la variante genética p.R375C eran significativamente menores que las generadas por canales no mutados.
ConclusionesEl fenotipo combinado de SSE, DAi y fenotipo de MCNC está asociado con la variante genética de HCN4 c.1123C> T (p.Arg375Cys). El gen HCN4 debería incluirse en el diagnóstico genético de la MCNC y en formas familiares de SSE, pero también en individuos con bradicardia sinusal y DAi.
Palabras clave
El canal regulado por nucleótidos cíclicos activado por hiperpolarización (HCN4) está constituido por tetrámeros que conforman los canales iónicos que conducen la corriente «funny» (If), que desempeña un papel fundamental en la actividad de marcapasos del nódulo sinoauricular. En 2003 se observó por primera vez la implicación de diferentes variantes de HCN4 con pérdida de función en las formas hereditarias del síndrome del seno enfermo (SSE)1. La evolución clínica de estos síndromes incluye arritmias supraventriculares (en particular fibrilación auricular [FA]), necesidad precoz de marcapasos, complicaciones cardioembólicas e incluso muerte súbita cardiaca2,3.
Recientemente, las variantes del gen HCN4 se han asociado con alteraciones estructurales cardiacas tales como miocardiopatía no compactada del ventrículo izquierdo (MCNC), prolapso de la válvula mitral y dilatación de la aorta ascendente4–6.
En este artículo se hace referencia a una gran familia con SSE hereditario asociado con dilatación de la aurícula izquierda (DAi) y MCNC causada por una variante rara de HCN4 (p.R375C), que muestra penetrancia completa y un curso clínico benigno.
MÉTODOSPruebas genéticasSe realizó al probando un estudio genético que incluyó 173 genes mediante tecnología de secuenciación de nueva generación (next-generation sequencing [NGS]) (Illumina HiSeq; Applied Biosystems 3730 DNA Analyzer, Thermo Fisher Inc., Estados Unidos). Se consideraron las variantes candidatas en función de los criterios de patogenicidad habituales7. El ADN se obtuvo a partir de muestras de sangre de adultos y de saliva de pacientes pediátricos. Se consideró que había segregación si una variante aparecía en todos los sujetos afectados y no en los sujetos sin el fenotipo. Se determinó cosegregación significativa con un valor discriminatorio de N1/328.
Cultivo celular y análisis electrofisiológico de la variante de elecciónLas corrientes de HCN4 (IHCN4) se registraron con la técnica de fijación de membranas en células de ovario de hámster chino transfectadas tanto con canales HCN4 no mutados (WT) como con canales mutados, tal como se ha descrito previamente9. Las densidades de corrientes se calcularon mediante la normalización de su amplitud en relación con la capacidad de la célula. Se analizó la dependencia del voltaje de la activación del canal HCN4 y se ajustaron las funciones de Boltzmann a los datos para calcular a qué potencial de membrana se activaban el 50% de los canales (Vh) y la pendiente (k) de la curva. La selectividad iónica de los canales HCN4 se evaluó midiendo las relaciones de densidad de la IHCN4 de los canales completamente activados.
Pacientes y evaluación clínicaLa evaluación clínica y el estudio genético de todos los miembros de la familia se ajustaron a los principios establecidos en la Declaración de Helsinki. Se informó a todos los pacientes de los objetivos de la investigación. En el caso de los menores, siempre se requirió el consentimiento de sus padres. Se evaluó a todos los sujetos en la unidad de cardiopatías familiares. El estudio inicial incluyó un electrocardiograma de 12 derivaciones y un ecocardiograma transtorácico.
Se consideró que los sujetos podían estar afectados si su frecuencia cardiaca (FC) era< 60 lpm (o en función del valor límite de referencia para la edad de los pacientes pediátricos10) o si mostraban hipertrabeculación/ausencia de compactación en la ecocardiografía transtorácica según los criterios estándar11,12. El estudio se completó con resonancia magnética cardiaca (RMC), Holter de 24 h y ergoespirometría junto con ecocardiografía transtorácica. Se diagnosticó MCNC a los pacientes que cumplían los criterios de Petersen o Jacquier13,14. Para los pacientes pediátricos, se individualizaron las indicaciones de pruebas complementarias.
Análisis estadísticoTodos los análisis estadísticos se realizaron con la versión 20.0.0 del software SPSS de IBM (IBM Corp., Estados Unidos). Las variables cualitativas se expresaron mediante número absoluto (n) y porcentaje (%) y las cuantitativas continuas, mediante la media±desviación estándar o la mediana (intervalo).
Los resultados del estudio funcional se expresaron con media±error estándar de la media y se compararon mediante la prueba de la t de Student, ANOVA seguida de prueba de Tukey o prueba de la U de Mann-Whitney, según su ajuste a la normalidad, así como con modelos de efectos mixtos combinados. Un valor de p < 0,05 se consideró estadísticamente significativo para todos los análisis. Pueden hallarse más detalles sobre la metodología en el .
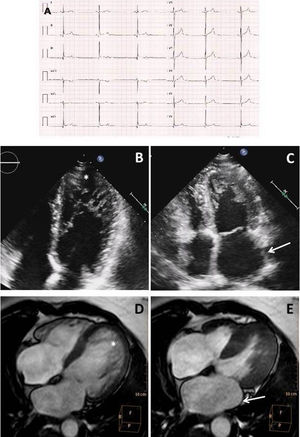
RESULTADOSEvaluación clínica del probandoEl probando era un varón de 18 años (III.19) con bradicardia sinusal, sin antecedentes clínicos de interés. El electrocardiograma en reposo mostró un intervalo QRS estrecho e intervalos PR y QT normales, con una elevación del punto J en las derivaciones inferiores (figura 1A). Los registros de Holter de ECG demostraron una FC mínima de 32 lpm y media de 44 lpm. La ergoespirometría objetivó una clase funcional excelente. La ecocardiografía transtorácica reveló DAi, dilatación del ventrículo izquierdo con hipertrabeculación y fracción de eyección normal (figura 1B,C). El estudio con RMC cumplía criterios diagnósticos de MCNC (figura 1D,E).
. C: foto en telesístole que muestra dilatación auricular izquierda (flecha). D: resonancia magnética cardiaca que muestra hipertrabeculación de ambos ventrículos (flechas) que cumple los criterios de miocardiopatía no compactada del ventrículo izquierdo. E: resonancia magnética cardiaca en telesístole que muestra dilatación auricular izquierda (flecha).")
Pruebas adicionales realizadas en el probando. A: electrocardiograma que indica bradicardia sinusal. B: ecocardiograma transtorácico apical de 4 cámaras que muestra hipertrabeculación profusa apical en el ventrículo izquierdo (asterisco). C: foto en telesístole que muestra dilatación auricular izquierda (flecha). D: resonancia magnética cardiaca que muestra hipertrabeculación de ambos ventrículos (flechas) que cumple los criterios de miocardiopatía no compactada del ventrículo izquierdo. E: resonancia magnética cardiaca en telesístole que muestra dilatación auricular izquierda (flecha).
No había antecedentes familiares de implante de marcapasos o muerte súbita cardiaca. Todos los familiares estaban asintomáticos en el momento de someterse a la evaluación inicial, excepto la madre del probando (II.10). Tenía antecedentes de FA paroxística y se había realizado con éxito una ablación de venas pulmonares. La abuela materna (I.2) murió a los 85 años debido a un ictus y el abuelo (I.1), a los 72 por una insuficiencia cardiaca de origen desconocido.
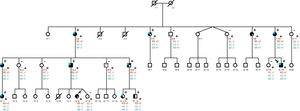
Se pudo evaluar a 22 sujetos: 2 familiares de primer grado (madre y hermana) y otros 20 de la línea materna (figura 2). En la primera evaluación, parecían afectados 12 familiares, todos ellos con bradicardia sinusal. Una prueba genética positiva lo confirmó después.
Resultados genéticos y análisis de la cosegregaciónEl análisis genético del probando identificó 3 variantes de significado incierto en heterocigosis: NM_005477.2:c.1123C>T;(p.R375C) en el gen HCN4, NM_001281740.1:c.1701C>A;(p.F567L) en el gen FHOD3 y NM_004415.2:c.4632G>T;(p.R1544S) en el gen DSP. De estas variantes, solo la c.1123C>T;(p.R375C) del gen HCN4 mostró segregación en la familia de acuerdo con el fenotipo. La variante p.R375C estaba registrada previamente (rs755356387) con una frecuencia alélica total de 0,000003977 (Base de Datos de Agregación de Genomas [gnomAD])15. La bradicardia sinusal mostró una penetrancia completa, incluso en los sujetos pediátricos. Así pues, teniendo en cuenta a los miembros de la familia afectados y no afectados, la bradicardia sinusal mostró una cosegregación muy fuerte (n=1/218). La penetrancia de hipertrabeculación/MCNC fue incompleta, con expresividad variable entre los sujetos, pero también mostró una cosegregación significativa (n=1/25). En este caso, para calcular la cosegregación solo se tuvo en cuenta a los sujetos afectados (figura 2). Las características principales de los portadores de la variante del gen HCN4 se muestran en la tabla 1 y la tabla 2.
: individuos estudiados por el equipo clínico. Símbolos tachados: personas fallecidas. Sombreados por la izquierda: bradicardia sinusal. Sombreados por la derecha: MCNC. Medio sombreados por la derecha: hipertrabeculación sin criterios de MCNC. Sombreado azul en la parte izquierda superior: dilatación auricular izquierda. G1: genotipo para HCN4:p.R375C; G2: genotipo para FHOD3:p.F567L; G3: genotipo para DSP:p.R1544S.+/–: heterocigosis; –/– homocigosis (no mutado). MCNC: miocardiopatía no compactada del ventrículo izquierdo.")
Árbol genealógico de la familia con la variante del gen HCN4. Los números que hay debajo del símbolo del sujeto indican la identificación del miembro de la familia. Cuadrados/círculos: varón/mujer. Asterisco (*): individuos estudiados por el equipo clínico. Símbolos tachados: personas fallecidas. Sombreados por la izquierda: bradicardia sinusal. Sombreados por la derecha: MCNC. Medio sombreados por la derecha: hipertrabeculación sin criterios de MCNC. Sombreado azul en la parte izquierda superior: dilatación auricular izquierda. G1: genotipo para HCN4:p.R375C; G2: genotipo para FHOD3:p.F567L; G3: genotipo para DSP:p.R1544S.+/–: heterocigosis; –/– homocigosis (no mutado). MCNC: miocardiopatía no compactada del ventrículo izquierdo.
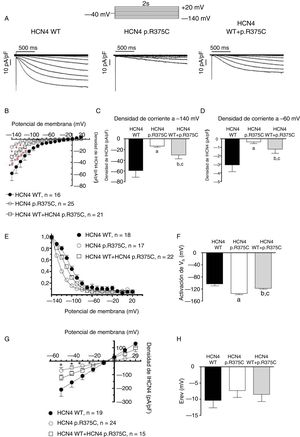
El recuadro de la izquierda de la figura 3A muestra el trazado de la IHCN4 generado tras aplicar pulsos de 2 s entre –140 y+20mV en fases de 10mV desde un potencial de retención de –40mV a células de ovario de hámster chino que expresan canales HCN4 WT. Los pulsos hiperpolarizantes generaron una corriente entrante que se activó lentamente hasta alcanzar un nivel estable, cuya amplitud disminuyó de manera progresiva a potenciales más positivos. Comparados con los canales WT, los canales HCN4 p.R375C generaron una corriente entrante menor y de activación más lenta (figura 3A, recuadro central). Teniendo en cuenta la situación de heterocigosis de los portadores, las células se cotransfectaron con los canales HCN4 WT y HCN4 p.R375C en una proporción 1:1. La amplitud de IHCN4 generada por la cotransfección fue inferior a la generada por los canales WT (figura 3A, recuadro de la derecha). Al analizar la densidad de IHCN4 en los distintos potenciales de membrana, se confirmó que las densidades de IHCN4 generadas por los canales p.R375C eran significativamente inferiores a las generadas por los canales WT+p.R375C y WT (n ≥ 16; p <0,01) (figura 3B). Además, la disminución de IHCN4 era evidente tanto en potenciales de membrana muy negativos (–140mV) (figura 3C) como fisiológicos (–60mV) (n ≥ 16; p <0,01) (figura 3D). Curiosamente, la densidad de la corriente generada por la cotransfección de los canales WT y p.R375C fue la mitad de la generada por los canales WT (n ≥ 16; p <0,01). Estos resultados indican que los canales p.R375C no actúan como «proteínas tóxicas» con un efecto negativo dominante.
 o –60mV (D). E: las densidades de la corriente de cola generadas por distintos canales tras aplicar pulsos de 1 s a –140mV se normalizaron y representaron frente al potencial de membrana del pulso de la prueba; las líneas continuas representan el ajuste de Boltzmann a los datos. F: el potencial de membrana que activa el 50% (Vh) de los canales WT, p.R375C y WT+p.R735C. G: relación densidad-voltaje de la IHCN4 totalmente activada generada por canales WT, p.R375C y WT+p.R375C; las líneas continuas representan la regresión lineal con respecto a los datos. H: Einv de los canales calculado a partir de la intersección de la regresión lineal con respecto a los datos con el eje de las abscisas de cada uno de los experimentos; de B a H, cada punto/barra representa la media±EEM de al menos 15 experimentos/células de al menos 3 placas. CHO: ovario de hámster chino; EEM: error estándar de la media; Einv: potencial inverso; IHCN4: corrientes de HCN4; WT: wild-type. ap <0,01 frente a HCN4 WT. bp <0,05 frente a HCN4 WT. cp <0,05 frente a p.R375C.")
Análisis electrofisiológico de la variante del gen HCN4. A: grupos de trazados de IHCN4 generados en células CHO que expresan de manera temporal canales HCN4 WT, p.R375C y WT+p.R375C mediante la aplicación del protocolo representado en la parte superior. B: relación densidad-voltaje generada en células que expresan canales WT, p.R375C y WT+p.R375C. C y D: densidad de IHCN4 generada por canales WT, p.R375C y WT+p.R375C con la aplicación de pulsos a –140 mV (C) o –60mV (D). E: las densidades de la corriente de cola generadas por distintos canales tras aplicar pulsos de 1 s a –140mV se normalizaron y representaron frente al potencial de membrana del pulso de la prueba; las líneas continuas representan el ajuste de Boltzmann a los datos. F: el potencial de membrana que activa el 50% (Vh) de los canales WT, p.R375C y WT+p.R735C. G: relación densidad-voltaje de la IHCN4 totalmente activada generada por canales WT, p.R375C y WT+p.R375C; las líneas continuas representan la regresión lineal con respecto a los datos. H: Einv de los canales calculado a partir de la intersección de la regresión lineal con respecto a los datos con el eje de las abscisas de cada uno de los experimentos; de B a H, cada punto/barra representa la media±EEM de al menos 15 experimentos/células de al menos 3 placas. CHO: ovario de hámster chino; EEM: error estándar de la media; Einv: potencial inverso; IHCN4: corrientes de HCN4; WT: wild-type. ap <0,01 frente a HCN4 WT. bp <0,05 frente a HCN4 WT. cp <0,05 frente a p.R375C.
Para cuantificar la cinética de activación, se ajustó una función monoexponencial a la activación del trazado generado por los pulsos a –130mV. Las constantes de tiempo de la activación de la IHCN4 (τ) se situaron en una media de 495±44, 1.960±387 y 727±85 ms en los canales HCN4 WT, HCN4 p.R375C y HCN4 p.R375C+WT respectivamente (n ≥ 16, p <0,01).
En la figura 3E se muestra un análisis de la activación de los canales HCN4 dependientes de voltaje (véase en el los registros patch-clamp). Los canales p.R375C se activaron a potenciales más negativos que en los canales WT, un efecto que explica probablemente la reducción de la densidad de la corriente producida por la variante. De hecho, Vh se hiperpolarizó de manera significativa (n ≥ 17; p <0,01) (figura 3F). La activación de los canales WT+p.R375C también se desplazó a potenciales negativos comparada con la de los canales WT (n ≥ 18; p <0,05) (figura 3E,F).
Para determinar si la variante altera la selectividad iónica de los canales HCN4, se determinó la relación entre las densidades de IHCN4 de los canales totalmente activados (figura 3G). Se calculó el potencial inverso (Einv) desde la intersección de la regresión lineal de los datos con el eje de las abscisas a concentraciones extracelulares e intracelulares de K+ de 30 y 142mM (véase en el los registros patch-clamp). El Einv no se modificó con la expresión solo de los canales p.R375C ni con la coexpresión con los canales WT (n ≥ 15; p> 0,05) (figuras 3G,H). Estos resultados indican que la variante no modificó la selectividad iónica de los canales HCN4.
Estudios adicionales en portadores de la variante de HCN4 y seguimientoEl individuo II.9 realizó su seguimiento clínico en otro centro médico, y no se dispone del estudio completo. Todos los portadores genéticos cumplían los criterios de disfunción sinusal y ninguno mostró FA en el electrocardiograma Holter (tabla 1). Curiosamente, IV.8 y IV.9 eran gemelas bivitelinas de 5 años; IV.9 (no portadora genética) tenía una FC en reposo de 100 lpm, en comparación con los 75 lpm de IV.8 (portadora de la variante HCN4).
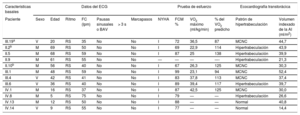
Características basales y electrocardiograma, prueba de esfuerzo y datos ecocardiográficos de la familia del portador de la variante p.R375C del gen HCN4
| Características basales | Datos del ECG | Prueba de esfuerzo | Ecocardiografía transtorácica | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Paciente | Sexo | Edad | Ritmo | FC (lpm) | Pausas sinusales> 3 s o BAV | Marcapasos | NYHA | FCM % | VO2 máximo (ml/kg/min) | % del VO2 predicho | Patrón de hipertrabeculación | Volumen indexado de la AI (ml/m2) |
| III.19a | V | 20 | RS | 35 | No | No | I | 72 | 36,5 | 87 | MCNC | 44,7 |
| II.2b | M | 69 | RS | 50 | No | No | I | 69 | 22,9 | 114 | Hipertrabeculación | 43,9 |
| II.5 | M | 68 | RS | 59 | No | No | I | 87 | 25 | 138 | Hipertrabeculación | 39,9 |
| II.9 | M | 61 | RS | 55 | No | No | — | — | — | —- | Hipertrabeculación | 21,3 |
| II.10b | M | 56 | RS | 40 | No | No | I | 67 | 26,3 | 125 | MCNC | 30,3 |
| III.1 | M | 48 | RS | 59 | No | No | I | 99 | 23,1 | 94 | MCNC | 52,4 |
| III.4 | V | 42 | RS | 41 | No | No | I | 83 | 37,8 | 113 | MCNC | 37,4 |
| III.6 | V | 36 | RS | 40 | No | No | I | 89 | 39,4 | 117 | Hipertrabeculación | 39,7 |
| IV.1 | M | 16 | RS | 37 | No | No | I | 87 | 42,5 | 125 | MCNC | 30,0 |
| IV.8 | M | 5 | RS | 75 | No | No | I | 79 | — | — | Hipertrabeculación | 26,6 |
| IV.13 | M | 12 | RS | 50 | No | No | I | 88 | — | — | Normal | 40,8 |
| IV.14 | V | 9 | RS | 55 | No | No | I | 77 | — | — | Normal | 14,4 |
AI: aurícula izquierda; BAV: bloqueo auriculoventricular; ECG: electrocardiograma; FC: frecuencia cardiaca; FCM: frecuencia cardiaca máxima; M: mujer; MCNC: miocardiopatía no compactada del ventrículo izquierdo; NYHA: clase funcional de la New York Heart Association; RS: ritmo sinusal; V: varón; VO2: consumo de oxígeno.
La ecocardiografía transtorácica demostró fenotipo de hipertrabeculación/MCNC en 10 de los 12 portadores genéticos (83%), DAi en 7 de los 12 portadores genéticos (58%) y un corazón normal en 2 sujetos pediátricos (tabla 1). No se observaron otras alteraciones estructurales en los individuos.
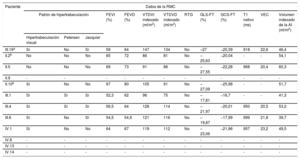
Se realizó RMC a 8 pacientes (se excluyó a los pacientes pediátricos) (tabla 2); 4 pacientes cumplieron los criterios diagnósticos de MCNC y otros 4 mostraron hipertrabeculación no patológica del ventrículo; 5 pacientes tenían dilatación moderada del ventrículo izquierdo y un tamaño normal del ventrículo derecho, y el probando (III.19) tenían una dilatación grave de ambos ventrículos. En 3 portadores de la variante HCN4 (III.1, III.4 y III.6) la FEVI era intermedia. En relación con los datos sobre deformación miocárdica, solo 1 paciente (III.1) presentó strain longitudinal global (GLS) y strain circunferencial global (GCS) patológicos. No se observó realce tardío de gadolinio en ningún paciente y en todos los valores de T1 nativo (965±15,8 ms) y volumen extracelular (21,7%±1,2%) fueron normales. Se observó DAi en todos los pacientes, grave en el 89%, y en todos la fracción de eyección auricular en la RMC fue normal (fracción de eyección, 56,6%; GLS, 35,8%).
Datos de la resonancia magnética cardiaca de la familia del portador de la variante p.R375C del gen HCN4
| Paciente | Datos de la RMC | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patrón de hipertrabeculación | FEVI (%) | FEVD (%) | VTDVI indexado (ml/m2) | VTDVD indexado (ml/m2) | RTG | GLS-FT (%) | GCS-FT (%) | T1 nativo (ms) | VEC | Volumen indexado de la AI (ml/m2) | |||
| Hipertrabeculación visual | Petersen | Jacquier | |||||||||||
| III.19a | Sí | No | Sí | 59 | 64 | 147 | 134 | No | –27 | −20,39 | 918 | 22,6 | 46,4 |
| II.2b | No | No | No | 65 | 72 | 86 | 81 | No | –25,63 | −20,04 | - | - | 54,1 |
| II.5 | No | No | No | 69 | 73 | 91 | 86 | No | –27,55 | −22,28 | 968 | 20,4 | 60,3 |
| II.9 | - | - | - | - | - | - | - | - | - | - | - | - | |
| II.10b | Sí | No | No | 67 | 80 | 105 | 91 | No | –27,09 | −25,98 | - | - | 51,7 |
| III.1 | Sí | Sí | Sí | 52,3 | 62 | 96 | 75 | No | –17,81 | −16,7 | - | - | 41,3 |
| III.4 | Sí | Sí | Sí | 56,5 | 64 | 128 | 114 | No | –21,97 | −20,01 | 950 | 20,5 | 53,2 |
| III.6 | Sí | No | Sí | 54,5 | 54,9 | 121 | 116 | No | –19,87 | −17,99 | 999 | 21,8 | 39,7 |
| IV.1 | Sí | No | No | 64 | 67 | 119 | 112 | No | –23,06 | −21,96 | 957 | 23,2 | 49,5 |
| IV.8 | - | - | - | - | - | - | - | - | - | - | - | - | - |
| IV.13 | - | - | - | - | - | - | - | - | - | - | - | - | - |
| IV.14 | - | - | - | - | - | - | - | - | - | - | - | - | - |
AI: aurícula izquierda; FEVD: fracción de eyección del ventrículo derecho; FEVI: fracción de eyección del ventrículo izquierdo; GCS-FT: strain circunferencial global mediante feature tracking; GLS-FT: strain longitudinal global mediante feature tracking; RMC: resonancia magnética cardiaca; RTG: realce tardío de gadolinio; VEC: volumen extracelular; VTDVD: volumen telediastólico del ventrículo derecho; VTDVI: volumen telediastólico del ventrículo izquierdo.
Se realizaron ergoespirometrías a todos los portadores genéticos adultos (tabla 1). Todos los portadores completaron la tercera fase del protocolo de Bruce y en 6 de ellos se observaron latidos ventriculares ectópicos. Los ecocardiogramas de esfuerzo mostraron una reserva contráctil positiva, sin otros indicadores patológicos. Todos los pacientes se hallaban en una clase funcional normal para el VO2 máximo teórico predicho (111%±15%), con un comportamiento normal del pulso de O2 (142%±30%) y VO2 en el umbral anaeróbico superior al predicho (72%±15%). Se evaluó mediante una prueba de esfuerzo convencional sobre cinta sin fin a los portadores pediátricos, que mostraron una clase funcional normal, sin arritmias significativas.
Al final de este estudio, todos los miembros de la familia estaban vivos (seguimiento medio, 29±9 meses) y en ninguno se había desarrollado insuficiencia cardiaca. Aparte del paciente con FA diagnosticada previamente, no se han documentado otras arritmias. En consecuencia, ningún paciente requirió marcapasos o desfibrilador automático implantable.
DISCUSIÓNEste estudio hace referencia a una familia extensa con un fenotipo combinado de bradicardia sinusal, DAi y MCNC debidas a la variante c.1123C>T;(p.R375C) del gen HCN4. Esta variante (RCV000693215.1; rs755356387) se había registrado previamente y se había descrito en un individuo con muerte súbita cardiaca y MCNC/bradicardia sinusal, pero no se realizó ningún estudio funcional ni de segregación familiar16. Según la recomendación consensuada del American College of Medical Genetics and Genomics7, se clasifica como «variante de significado incierto».
Las variantes de HCN4 se relacionaron inicialmente con SSE hereditario sin cardiopatía estructural2. En 2014, se describió por primera vez la relación entre MCNC y el SSE en varias familias con variantes en el gen HCN44,5. Hasta ahora, solo se han descrito 68 casos en 16 familias distintas. La presencia de hipertrabeculación del ventrículo izquierdo/MCNC y distintos grados de disfunción del nódulo sinusal, así como el síndrome de taquicardia-bradicardia con FA17, ha sido constante en todos los casos descritos y en ocasiones se ha vinculado a otros trastornos, como el prolapso de la válvula mitral4,5 o la dilatación de la aorta ascendente6. La necesidad de implante de marcapasos parece que es elevada en este grupo de pacientes y, entre todos los casos descritos, 4 sufrieron una muerte súbita cardiaca recuperada por fibrilación ventricular y 1 paciente falleció por muerte súbita cardiaca5,16,18.
Consideramos que la patogenicidad de la variante c.1123C>T;(p.R375C) del gen HCN4 está demostrada según los criterios de referencia7. Los estudios familiares muestran un patrón de herencia tipo autosómico dominante con penetrancia completa en la bradicardia sinusal e incompleta, pero igualmente elevada, en la DAi y la ausencia de compactación8,17. La gran penetrancia de la bradicardia sinusal se había descrito previamente en sujetos pediátricos4,17 y podría considerarse un marcador de la enfermedad. Los portadores de la variante c.1123C>T;(p.R375C) del gen HCN4 de la familia en estudio mostraron un curso benigno comparado con estudios previos16. La única complicación identificada durante el seguimiento fue un episodio de FA persistente, con un excelente control del ritmo con la ablación de las venas pulmonares.
Mecanismos de las variantes del gen HCN4 causantes del SSELos resultados muestran que la variante p.R375C modifica profundamente la dependencia de voltaje y la cinética de activación del canal HCN4. Esto redujo de un modo significativo la disponibilidad del canal y disminuyó la densidad de la corriente generada. Se ha demostrado con anterioridad la causalidad de las variantes del HCN4 mediante análisis electrofisiológico celulares4,5,17–19. Los canales HCN4 mutados no eran funcionales, lo cual resultó en una reducción de la corriente If. Los datos electrofisiológicos del presente estudio proporcionaron pruebas sólidas de la causalidad de la variante p.R375C en la patogenia de la bradicardia sinusal observada.
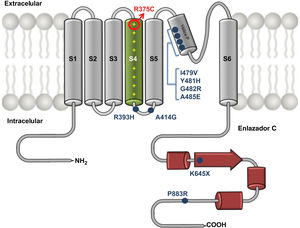
El residuo p.R375C se localiza en el segmento S4 de los canales HCN4 y es uno de los 7 residuos de este dominio del canal cargados positivamente (figura 4). De hecho, el segmento S4 es el sensor de voltaje del canal y la variante sustituye uno de los residuos de Arg cargados positivamente por un residuo polar neutro (Cys). Esto explica a la perfección los cambios profundos producidos por la variante en la dependencia de voltaje y la cinética de la activación del canal. Anteriormente se describieron resultados parecidos para la variante p.R378C observada en un paciente con SSE20.
. Ilustración de la estructura del canal HCN4 que representa las variantes que combinan MCNC y el fenotipo de SSE descritos hasta la actualidad. La Arg375 (círculo) se localiza en el segmento S4 y es uno de los 7 residuos cargados positivamente de este dominio que constituye el sensor de voltaje del canal. MCNC: miocardiopatía no compactada del ventrículo izquierdo; SSE: síndrome del seno enfermo.")
Esquema de un canal mutado HCN4 c.1123C>T;(p.R375C). Ilustración de la estructura del canal HCN4 que representa las variantes que combinan MCNC y el fenotipo de SSE descritos hasta la actualidad. La Arg375 (círculo) se localiza en el segmento S4 y es uno de los 7 residuos cargados positivamente de este dominio que constituye el sensor de voltaje del canal. MCNC: miocardiopatía no compactada del ventrículo izquierdo; SSE: síndrome del seno enfermo.
La MCNC se ha relacionado con distintas variantes que afectan a los genes del sarcómero, del citoesqueleto y de la membrana nuclear. Los modelos animales indican que la hipertrabeculación se debe a una alteración en la regulación de la proliferación, la diferenciación y la maduración celulares durante la formación de la pared del ventrículo21.
Hasta ahora, se conocen como mínimo 8 variantes distintas del gen HCN4 relacionadas con este fenotipo combinado (P883R, K645X, A485E, G482R, Y481H, I479V, A414G, R393H)4,5,17–19. Podría haber una relación entre el desarrollo del sistema de conducción cardiaco y la maduración del miocardio que explicaría la concomitancia de miocardiopatía y arritmias específicas22. Estas variantes del HCN4 podrían considerarse la «vía común final» que producen el fenotipo mixto23, pero todavía no se ha demostrado de qué modo se desarrolla la hipertrabeculación. Otra hipótesis sobre la aparición de la MCNC es que se trate de un rasgo adquirido de remodelación adaptativa en respuesta a la bradicardia sinusal5. En estudios previos, se ha descrito que los genes que codifican canales iónicos (SCN5A y RYR2) participan en la fisiopatología de la miocardiopatía24,25. En este estudio, se hallaron algunos datos confusos sobre si la familia tiene un rasgo fenotípico adquirido o una miocardiopatía real. En casi todos los portadores, los resultados de la FEVI y la deformación del miocardio fueron normales o intermedios, así como la clase funcional y la reserva contráctil positiva. Ninguno de los sujetos analizados presentó realce tardío de gadolinio ni alteraciones en el volumen extracelular o los mapas en T1.
Curiosamente, se observó una gran penetrancia de DAi con contractilidad auricular normal. Este hallazgo no se había relacionado previamente con las variantes del gen HCN4. Se desconoce cuál es el mecanismo causante de la DAi. Tal como se ha explicado previamente, la DAi podría ser resultado de una alteración real de la estructura anatómica auricular, ya que el HCN4 contribuye al desarrollo embrionario del ventrículo izquierdo y ambas aurículas26, o podría ser una adaptación fisiológica a la bradicardia sinusal.
Variantes del gen HCN4 con implicaciones en el SSE, la DAi y la MCNCLa relación entre las variantes del gen HCN4 y el fenotipo combinado del SSE, la DAi y la MCNC es innegable. El patrón sin compactación mostrado por los pacientes con la variante p.R378C del gen HCN4 indica poca agresividad y buen pronóstico. La remodelación de la aurícula izquierda es un importante sustrato para la FA y podría relacionarse con la aparición de la FA hereditaria, ya asociada con las variantes del gen HCN417.
Actualmente no están establecidos la evaluación y el seguimiento de los pacientes con variantes emergentes del gen HCN4. El seguimiento que se propuso para esta familia tiene por objetivo principal detectar complicaciones arrítmicas, principalmente en los fenotipos con DAi, con una alta probabilidad de aparición de FA, y disfunción ventricular sistólica. Se decidió llevar a cabo una revisión clínica anual con electrocardiograma y electrocardiograma Holter. El ecocardiograma se repetirá cada 1 o 2 años (excepto por cambios en la situación clínica).
Limitaciones del estudioEste estudio incluyó únicamente a una familia y no fue posible evaluar a todos sus miembros. No obstante, se ha evaluado a un gran número de ellos. De hecho, este estudio hace referencia a la mayor familia relacionada con el gen HCN4 descrita hasta la fecha, evaluada mediante un estudio clínico exhaustivo que incluyó ergoespirometría y RMC.
Como en los casos descritos previamente, nuestro trabajo no explica el mecanismo del efecto de la variante del gen HCN4 en la aparición de hipertrabeculación del ventrículo izquierdo o DAi. Sin embargo, este es el primer trabajo en que se examinó a los pacientes mediante RMC con técnicas paramétricas y deformación miocárdica, y se ha demostrado la existencia de una posible disfunción sistólica subclínica y DAi con posibles implicaciones clínicas.
CONCLUSIONESEl fenotipo combinado de SSE, DAi y MCNC se relaciona con distintas variantes hereditarias del gen HCN4. En los pacientes con formas hereditarias del SSE, se debería descartar una posible cardiopatía estructural, y las variantes del gen HCN4 deberían incluirse en el diagnóstico genético, incluso de los sujetos con bradicardia sinusal y DAi solas. Además, las variantes del gen HCN4 deberían añadirse a otros genes reconocidos en el estudio de los pacientes con MCNC.
Se describe a una familia con una variante c.1123C>T;(p.R375C) del gen HCN4 causante del fenotipo combinado de SSE, DAi y MCNC con un curso benigno.
Se requieren otros estudios para explicar el mecanismo fisiopatológico de la dilatación auricular y el fenotipo de hipertrabeculación/ausencia de compactación, así como para caracterizar la evolución natural de los pacientes afectados por las variantes HCN4 y la estratificación del riesgo en esta población específica.
- –
La relación entre las variantes del gen HCN4 y el fenotipo combinado de SSE, DAi y MCNC es innegable con la nueva evidencia, y con fenotipos que pueden variar según la variante subyacente.
- –
Las principales complicaciones relacionadas con las variantes del gen HCN4 son las complicaciones arrítmicas, como la FA y la muerte súbita, la necesidad precoz de marcapasos, las complicaciones cardioembólicas y la disfunción ventricular sistólica.
- –
Se observó una gran penetrancia de la DAi con contractilidad auricular normal. Este hallazgo no se había descrito previamente asociado con variantes del gen HCN4. La importancia de este hallazgo es que esta alteración estructural podría relacionarse con la aparición de FA hereditaria, previamente relacionada con las variantes del gen HCN4.
- –
El patrón sin compactación mostrado por los pacientes con la variante p.R378C del gen HCN4 indica poca agresividad y buen pronóstico.
- –
El seguimiento propuesto para estos pacientes con variantes de HCN4 tiene por objetivo principal detectar complicaciones arrítmicas, principalmente en los fenotipos con DAi, con una alta probabilidad de aparición de FA, y disfunción ventricular sistólica.
El presente estudio fue financiado en parte por el Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares (CIBERCV), con el apoyo del Instituto de Salud Carlos III, la Comunidad de Madrid (B2017/BMD-3738) y el Ministerio de Economía y Competitividad (SAF2017-88116-P) para R. Caballero y E. Delpón. M. Alonso-Fernández-Gatta cuenta con la financiación de un contrato Río Hortega (CM19/00055) con el apoyo del Instituto de Salud Carlos III (cofinanciado por el Fondo Social Europeo «FSE Invierte en tu futuro»).
CONFLICTO DE INTERESESLos autores no tienen ningún conflicto de intereses que declarar.
Se puede consultar material adicional a este artículo en su versión electrónica disponible en https://doi.org/10.1016/j.recesp.2020.06.037