

La evaluación del riesgo en la hipertensión arterial pulmonar (HAP) es esencial de cara a administrar un tratamiento más agresivo a aquellos pacientes de mayor riesgo. Sin embargo, las escalas pronósticas más recientes olvidan el trasfondo genético. Además, la enfermedad venooclusiva pulmonar (EVOP) no se ha considerado nunca en las estrategias de evaluación del riesgo.
MétodosSe consideraron para este trabajo pacientes consecutivos incluidos en el registro Español de HAP (REHAP) analizados genéticamente entre los años 2011 y 2022. Se aplicó en ellos el modelo COMPERA 2.0 de 4 estratos, comparando este resultado con el obtenido de un modelo ampliado que incluyó la genética. Se usaron modelos de regresión de Cox y el estadístico C de Harrel para comparar los distintos modelos. Se estudiaron específicamente estos modelos en la población EVOP antes de su inclusión.
ResultadosSe seleccionaron 298 pacientes con HAP idiopática, familiar, inducida por fármacos y EVOP del registro REHAP. Considerando únicamente aquellos con todas las variables de interés disponibles al diagnóstico (clase funcional, prueba de la marcha de los 6 minutos, NT-proBNP o BNP), e incluidos en el modelo de 4 estratos (n=142), después de una mediana de seguimiento de 58,2 meses hasta el 17,6% de los pacientes fallecieron y un 11,3% necesitaron trasplante pulmonar. La aplicación del modelo de 4 estratos en nuestra población demostró una buena capacidad pronóstica (C de Harrel de 0,689). La introducción de la genética no mejoró ésta (índice C de 0,690). Este último modelo ampliado mostró una tendencia a una mejor identificación de pacientes en riesgo intermedio-bajo e intermedio-alto, sin diferencias en la identificación entre los estratos de riesgo intermedio-alto y alto.
ConclusionesEn este trabajo la adición del resultado del estudio genético al modelo de 4 estratos COMPERA alcanzó una capacidad pronóstica total similar al modelo original, pero cambió la identificación de los estratos de riesgo en una cohorte de pacientes jóvenes analizados genéticamente.
Palabras clave
La hipertensión arterial pulmonar (HAP) es una enfermedad grave y muy poco frecuente. Con independencia de cuál sea su causa, la suma de la disfunción endotelial, la inflamación, la vasoconstricción y la proliferación celular conducen en última instancia a un aumento de las presiones pulmonares y el consiguiente fracaso ventricular derecho1. Numerosas investigaciones realizadas en este campo a lo largo de las 2 últimas décadas han relacionado las alteraciones de ciertas vías moleculares que intervienen en el estrés oxidativo, la inflamación o la señalización celular con la aparición de esta enfermedad, entre otras2. Se han observado variantes del gen que codifica el receptor de la proteína morfogenética ósea de tipo II (BMPR2) en hasta un 86% de los casos familiares y en entre un 14% y un 35% de los casos esporádicos. Otros genes implicados en la función endotelial pulmonar se han asociado también con el desarrollo de HAP3. Además, la enfermedad venooclusiva pulmonar (EVOP) es una forma especialmente infrecuente y extremadamente agresiva de HAP. Su forma heredable es causada por variantes homocigotas en el gen que codifica el factor de iniciación de la traducción eucariota 2 alfacinasa 4 (EIF2AK4)4 y suele estar infradetectada y catalogada en alguna ocasión como forma idiopática5. Tanto la presencia de variantes genéticas en el gen BMPR2 como la EVOP son factores de mal pronóstico en esta enfermedad6,7.
A pesar de las mejoras sustanciales en la supervivencia que se produjeron después de la introducción de los vasodilatadores pulmonares específicos, el pronóstico de la HAP continúa siendo muy desfavorable. La evaluación del riesgo individual es esencial para proporcionar un tratamiento más agresivo a los pacientes con un mayor riesgo de muerte8. En los últimos años se han diseñado múltiples escalas pronósticas sencillas, con resultados comparables entre ellas9,10. No obstante, ninguna de estas escalas incluyen el trasfondo genético ni pacientes con EVOP. En este trabajo sugerimos, no solo incluir las bases moleculares para la clasificación correcta de los pacientes con HAP, sino también la necesidad de evaluar sistemáticamente las bases moleculares en los pacientes con HAP/EVOP, lo cual podría mejorar la estratificación pronóstico gracias a la genética.
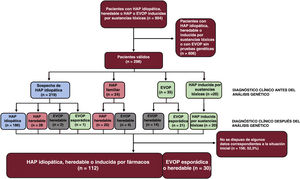
MÉTODOSPoblación del estudioEl Registro Español de Hipertensión Arterial Pulmonar (REHAP) es un registro prospectivo que se inició en enero de 200711. Para la inclusión en el registro, el diagnóstico de la HAP requirió un cateterismo cardíaco derecho, con una presión arterial pulmonar media ≥ 25mmHg, una resistencia vascular pulmonar ≥ 3 UW y una presión arterial pulmonar enclavada ≤ 15mmHg. En este estudio incluimos a pacientes de edad superior a 18 años con HAP idiopática, heredable o inducida por drogas o toxinas, y también a los que presentaban una EVOP esporádica o heredable. El estudio genético se ofreció a todos los pacientes con HAP idiopática, familiar o asociada a fármacos o con una EVOP. Se seleccionó tan solo a los pacientes con un resultado definitivo en una prueba genética. El período de estudio se inició en 2011 y finalizó en febrero de 2021.
La EVOP se confirmó si había una variante homocigota en el gen EIF2AK4, o tras la confirmación histológica. Esta enfermedad se diagnosticó también si había un deterioro respiratorio después del inicio del tratamiento con vasodilatadores pulmonares. De igual modo, el trastorno se consideró probable si había una capacidad de difusión pulmonar baja para el monóxido de carbono (DLCO) y cuando se daban como mínimo 2 signos radiológicos de EVOP de los 3 signos posibles12.
Se obtuvo el consentimiento informado por escrito en todos los casos. Los datos del REHAP y los estudios genéticos siguieron los principios de la Declaración de Helsinki. El protocolo fue aprobado por los comités de ética de investigación de todos los centros participantes. Los datos de las variables clínicas, demográficas, analíticas, funcionales y hemodinámicas se obtuvieron del registro REHAP.
Análisis genéticosEl estudio molecular en la HAP se inició en 2011 mediante secuenciación Sanger y amplificación de sondas dependiente de ligadura múltiple (MLPA) en los genes BMPR2, TBX4 y KCNK3. Este análisis se amplió de 2014 a 2020 mediante un panel de secuenciación de nueva generación (NGS) de 21 genes (HAP v 1.2), que se amplió hasta abarcar 35 genes (HAP v 3) sobre la base de datos de investigación previos. El panel se diseñó con NimbleDesign (Roche, Estados Unidos). La fragmentación y la preparación de la biblioteca se llevaron a cabo con el SeqCap EZ Choice Enrichment Kit (Roche, Estados Unidos) y la secuenciación se realizó con las plataformas Illumina MiSeq y NextSeq500 (Illumina, Estados Unidos). La priorización de variantes se detalla en la y se aplicó un script propio desarrollado internamente para analizar las variantes de número de copias13.
Posteriormente, en 2020, pasamos a la tecnología de secuenciación de exoma completo. La preparación de la biblioteca se llevó a cabo mediante Agilent SureSelect TM (v 6.0) y todos los kits de exones fueron seguidos de una secuenciación en un NovaSeq Sequencer (Illumina, Estados Unidos). La priorización de variantes se realizó también mediante VarSeq (Golden Helix, Estados Unidos) (figura 1). Después de la priorización de variantes, las variantes candidatas se clasificaron según las directrices del American College of Medical Genetics14.

Se proporcionó un asesoramiento genético a cada uno de los pacientes incluidos y a sus familiares de primer grado. Aunque se registraron los antecedentes familiares durante el primer estudio genético, se obtuvo una muestra de ADN para el análisis únicamente de los probandos. Cuando se detectaba una variante genética, se ofrecía el estudio a los familiares de primer grado. En consecuencia, se llevaron a cabo análisis de segregación en esos casos, reclasificando la variable según los resultados de estos análisis y según la presencia de otros factores de patogenia, según las directrices del American College of Medical Genetics. Se buscó la posible presencia de hipertensión pulmonar en los familiares de primer grado con un resultado genético positivo, utilizando para ello la ecocardiografía transtorácica, el electrocardiograma y la exploración física. En los familiares de primer grado de los pacientes con EVOP, se determinó también la capacidad de difusión pulmonar para el monóxido de carbono.
ResultadosEl objetivo principal fue el primer episodio de trasplante de pulmón o la muerte. Se elaboró un modelo de regresión de Cox con la inclusión de las variables del modelo de 4 estratos validado propuesto por el registro COMPERA y validado por el registro de hipertensión pulmonar francés (FPHR)9,15. El modelo de 4 estratos original se comparó con un modelo que incluía las mismas 3 variables originales y además las pruebas genéticas. Esta última variable se consideró positiva si había como mínimo 1 variante patógena (P) o probablemente patogénica (PP). Se tomó como tiempo cero la fecha del primer cateterismo cardíaco derecho. La utilidad del modelo que incluye la EVOP se evaluó específicamente en esta cohorte antes de su inclusión en la población global del estudio. Los valores de corte para los modelos de 4 estratos fueron los mismos que se han definido anteriormente9. Para introducir la genética en el modelo COMPERA 2.0, comparamos el coeficiente obtenido para esta variante en el análisis de regresión de Cox univariante con el valor medio de los coeficientes obtenidos para cada una de las demás variables del modelo de 4 estratos original, de manera similar al método utilizado en el estudio REVEAL original16. En consecuencia, consideramos que a la ausencia de una variable genética se le asignaba 1 punto y a su presencia se le asignaban 3 puntos en el modelo ampliado que incluía la genética ().
Análisis estadísticoLas variables cualitativas se presentan mediante la frecuencia absoluta y relativa, y se compararon con la prueba exacta de Fisher o la prueba de Pearson, según procediera. Las variables continuas se presentan en forma de media±desviación estándar o de mediana [rango intercuartílico] y se compararon con pruebas de t o pruebas de Wilcoxon según procediera. Las curvas de supervivencia de Kaplan-Meier se compararon con la prueba del logaritmo del rango (log-rank). Los modelos de regresión de Cox se compararon mediante los resultados del estadístico C de Harrel, utilizando la prueba de D de Somers, con la inclusión de tan solo los pacientes en los que se disponía de todas las variables de interés (clase funcional de la Organización Mundial de la Salud, prueba de la marcha de 6 minutos, péptido natriurético tipo B o propéptido natriurético aminoterminal tipo B) en la situación inicial. No se realizaron imputaciones de los datos no disponibles. Para el análisis de los datos se utilizó el programa Stata versión 14.0 (StataCorp, College Station, Estados Unidos) y el programa R studio (v 4.0.3).
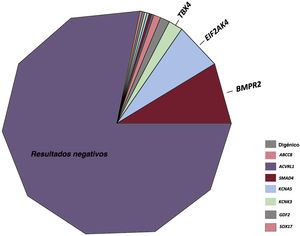
RESULTADOSResultados de las pruebas genéticas en el registro REHAPEntre enero de 2011 y febrero de 2021, identificamos a 298 pacientes con pruebas genéticas definitivas en el grupo de pacientes con HAP idiopática, familiar, inducida por fármacos o EVOP (figura 1). Este grupo representaba un 33,0% del número total de individuos que estuvieron en seguimiento activo en el registro REHAP durante este periodo (298 de los 904 pacientes del registro con esos criterios de inclusión). Las características iniciales de los pacientes incluidos pueden observarse en la . En este grupo, en hasta un 49,4% de los pacientes sin resultado genético positivo no se realizó una determinación de los biomarcadores cardíacos en la situación inicial. No se dispuso de esta variable en el 39,5% del grupo de los pacientes con una variante P o PP. Se dispuso de información sobre la distancia recorrida en la prueba de la marcha de 6 minutos en más del 92% de los pacientes de ambos grupos, y se pudo determinar la clase funcional de todos los participantes (). Después de los análisis genéticos, hubo 65 pacientes que tenían como mínimo una variante P o PP (un 21,8% de la cohorte). Se identificó una variante de significado incierto como única variante genética en 22 pacientes (un 7,4% de la cohorte total). En el grupo de pacientes que tenían como mínimo una variante genética P o PP, la distribución de las variantes genéticas fue la siguiente: BMPR2 (26 casos), variantes digénicas incluida una P o PP en el BMPR2 (4 casos), EIF2AK4 (20 casos), TBX4 (6 casos), ABCC8 (3 casos) y otros genes (6 casos). En el grupo de pacientes con una variante de significado incierto, las variantes más frecuentes se presentaron en ABCC8 (5 casos) y en NOTCH3 (4 casos). También hubo variantes en los siguientes genes: BMPR1B, CAV1 (x 2), CBLN2, CPS1, CYP1A1, EIF2AK4, ENG, GDF2, KCNA5 (x 2), KCNK3 y SMAD9 clasificadas como variantes de significado incierto ().
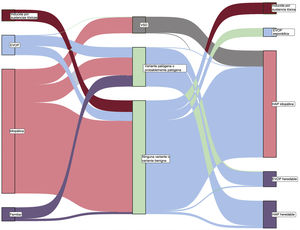
Teniendo en cuenta el trasfondo clínico, 219 casos se clasificaron inicialmente como HAP idiopática. Después del análisis genético, 31 casos fueron reclasificados como otros subtipos de hipertensión pulmonar (14,2%), la mayor parte de ellos reclasificados como HAP heredable debido a la presencia de una variante P o PP. De los 24 casos inicialmente clasificados como de HAP familiar, en la inmensa mayoría se confirmó una clasificación de HAP heredable (83,3%), pero 4 casos fueron reclasificados como EVOP heredable, debido al descubrimiento de una variante en homocigosis en el gen EIF2AK4 (16,7%). Por último, en los 41 pacientes con una sospecha clínica de EVOP, las pruebas genéticas permitieron la reclasificación y el diagnóstico de EVOP en 14 de ellos (34,1%) (figura 2 y figura 3).

Diagrama de Sankey que muestra los resultados de las pruebas genéticas y la reclasificación de la forma de HAP en función del resultado del análisis molecular. HAP, hipertensión arterial pulmonar; HP, hipertensión pulmonar; EVOP, enfermedad venooclusiva pulmonar; VSD, variante de significado desconocido.

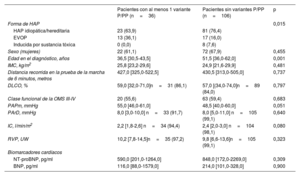
Al considerar tan solo los pacientes en los que se disponía de todas las variables en la situación inicial y que fueron incluidos en el modelo de 4 estratos (n=142), la mediana de edad fue de 46,0 años (34,0-59,0). En comparación con los pacientes sin variantes genéticas significativas (n=106), los pacientes con una variante P o PP eran de una edad significativamente inferior (36,5 frente a 51,5 años; p=0,001). Además, los pacientes con variantes P o PP tenían unos parámetros hemodinámicos ligeramente peores (tabla 1). La prevalencia de la EVOP fue comparativamente mayor en el grupo de pacientes con variantes genéticas P o PP. En comparación con los pacientes con una EVOP esporádica, los que tenían EVOP heredable mostraron una edad de diagnóstico más temprana, una mejor clase funcional y concentraciones inferiores de los biomarcadores cardiacos, pero la gravedad hemodinámica fue similar ().
Características iniciales de los pacientes incluidos
| Pacientes con al menos 1 variante P/PP (n=36) | Pacientes sin variantes P/PP (n=106) | p | |
|---|---|---|---|
| Forma de HAP | 0,015 | ||
| HAP idiopática/hereditaria | 23 (63,9) | 81 (76,4) | |
| EVOP | 13 (36,1) | 17 (16,0) | |
| Inducida por sustancia tóxica | 0 (0,0) | 8 (7,6) | |
| Sexo (mujeres) | 22 (61,1) | 72 (67,9) | 0,455 |
| Edad en el diagnóstico, años | 36,5 [30,5-43,5] | 51,5 [36,0-62,0] | 0,001 |
| IMC, kg/m2 | 25,8 [23,2-29,6] | 24,9 [21,6-29,9] | 0,481 |
| Distancia recorrida en la prueba de la marcha de 6 minutos, metros | 427,0 [325,0-522,5] | 430,5 [313,0-505,0] | 0,737 |
| DLCO, % | 59,0 [32,0-71,0]n=31 (86,1) | 57,0 [(34,0-74,0]n=89 (84,0) | 0,797 |
| Clase funcional de la OMS III-IV | 20 (55,6) | 63 (59,4) | 0,683 |
| PAPm, mmHg | 55,0 [46,0-61,0] | 48,5 [40,0-60,0] | 0,051 |
| PArD, mmHg | 8,0 [3,0-10,0] n=33 (91,7) | 8,0 [5,0-11,0] n=105 (99,1) | 0,640 |
| IC, l/min/m2 | 2,2 [1,8-2,6] n=34 (94,4) | 2,4 [2,0-3,0] n=104 (98,1) | 0,080 |
| RVP, UW | 10,2 [7,8-14,5]n=35 (97,2) | 9,8 [6,6-13,6]n=105 (99,1) | 0,323 |
| Biomarcadores cardíacos | |||
| NT-proBNP, pg/ml | 590,0 [201,0-1264,0] | 848,0 [172,0-2269,0] | 0,309 |
| BNP, pg/ml | 116,0 [88,0-1579,0] | 214,0 [101,0-328,0] | 0,900 |
IMC, índice de masa corporal; IC, índice cardiaco; DLCO, capacidad de difusión pulmonar de monóxido de carbono; PP, probablemente patógena; PAPm, presión arterial pulmonar media; NT-proBNP, propéptido natriurético tipo B aminoterminal; P, patógena; HAP, hipertensión arterial pulmonar; EVOP, enfermedad venooclusiva pulmonar; RVP, resistencia vascular pulmonar; PArD, presión auricular derecha; OMS, Organización Mundial de la Salud.
Los datos se expresan en forma de número (%) o mediana [rango intercuartílico].
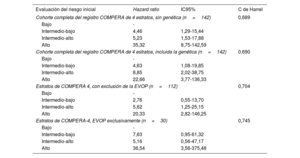
Después de una mediana de seguimiento de 58,2 [31,4-90,7] meses, el 17,6% de los pacientes había fallecido y un 11,3% de los pacientes había recibido trasplante bipulmonar. Al examinar específicamente la EVOP, la aplicación del modelo COMPERA de 4 estratos mostró en esa cohorte una bondad de ajuste similar a la obtenida en el resto de la población del estudio (estadístico C de Harrel de 0,745 en la EVOP en comparación con 0,704 en la cohorte total con la exclusión de la EVOP) (tabla 2). Globalmente, la aplicación de los criterios de la escala de riesgo de 4 estratos en nuestra población puso de manifiesto una buena capacidad pronóstica (estadístico C de Harrel de 0,689). La introducción del resultado genético no alcanzó una mejor capacidad pronóstica según lo evaluado con el estadístico C (índice C de 0,690 para el modelo ampliado con la adición de la genética, p=0,367). El uso de la genética mostró una tendencia no significativa a la identificación de los pacientes con un riesgo intermedio-bajo e intermedio-alto (p=0,053) (figura 4). El modelo que incluyó la genética no mostró diferencias estadísticamente significativas entre los pacientes en riesgo intermedio-alto y los de riesgo alto (p=0,114 con el modelo que incluía la genética frente a p <0,001 en el modelo original) (figura 4; tabla 2 y ).
Modelos de regresión de Cox de los pacientes incluidos
| Evaluación del riesgo inicial | Hazard ratio | IC95% | C de Harrel |
|---|---|---|---|
| Cohorte completa del registro COMPERA de 4 estratos, sin genética (n=142) | 0,689 | ||
| Bajo | - | ||
| Intermedio-bajo | 4,46 | 1,29-15,44 | |
| Intermedio-alto | 5,23 | 1,53-17,88 | |
| Alto | 35,32 | 8,75-142,59 | |
| Cohorte completa del registro COMPERA de 4 estratos, incluida la genética (n=142) | 0.690 | ||
| Bajo | - | ||
| Intermedio-bajo | 4,63 | 1,08-19,85 | |
| Intermedio-alto | 8,85 | 2,02-38,75 | |
| Alto | 22,66 | 3,77-136,33 | |
| Estratos de COMPERA 4, con exclusión de la EVOP (n=112) | 0,704 | ||
| Bajo | - | ||
| Intermedio-bajo | 2,76 | 0,55-13,70 | |
| Intermedio-alto | 5,62 | 1,25-25,15 | |
| Alto | 20,33 | 2,82-146,25 | |
| Estratos de COMPERA-4, EVOP exclusivamente (n=30) | 0,745 | ||
| Bajo | - | ||
| Intermedio-bajo | 7,63 | 0,95-61,32 | |
| Intermedio-alto | 5,16 | 0,56-47,17 | |
| Alto | 36,54 | 3,56-375,48 | |
IC95%, intervalo de confianza del 95%; EVOP, enfermedad venooclusiva pulmonar.
 e incluyendo la genética (B). Prueba de orden logarítmico (log-rank) p <0,001 para ambos modelos.")
La identificación de una variante patogénica o probablemente patogénica en genes relacionados con la aparición de HAP o EVOP se asoció a múltiples reclasificaciones de un grupo de HAP a otro. En una cohorte de pacientes jóvenes con HAP o EVOP, el modelo COMPERA de 4 estratos no mostró una buena diferenciación de los pacientes en riesgo intermedio-bajo y riesgo intermedio-alto. La inclusión del trasfondo genético mostró una tendencia a una mejor discriminación de los pacientes en riesgo intermedio (figura 5).
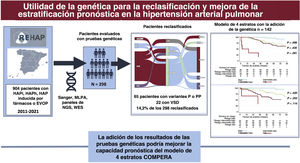
 con los casos de HAP idiopática, heredable o inducida por fármacos, así como los de EVOP, durante el período 2011-2021. Se realizaron pruebas genéticas en un total de 298 pacientes. Se encontraron variantes P o PP en 65 de estos 298 pacientes y, como consecuencia de ello, se reclasificó clínicamente al 14,2% de ellos. La adición del resultado de las pruebas genéticas no mejoró la capacidad pronóstica global del modelo de 4 estratos basado en el registro COMPERA 2.0. HAPh, hipertensión arterial pulmonar heredable; HAPi, hipertensión arterial pulmonar idiopática; NGS, secuenciación de nueva generación; PP, variante probablemente patógena; P, variante patógena; HAP, hipertensión arterial pulmonar; HP, hipertensión pulmonar; EVOP, enfermedad venooclusiva pulmonar; VSD, variante de significado desconocido; WES, secuenciación de exoma completo.")
Figura central. En la figura se resumen los principales resultados del estudio. En el recuadro de la izquierda se muestra un resumen de la población total del registro español de hipertensión pulmonar (REHAP) con los casos de HAP idiopática, heredable o inducida por fármacos, así como los de EVOP, durante el período 2011-2021. Se realizaron pruebas genéticas en un total de 298 pacientes. Se encontraron variantes P o PP en 65 de estos 298 pacientes y, como consecuencia de ello, se reclasificó clínicamente al 14,2% de ellos. La adición del resultado de las pruebas genéticas no mejoró la capacidad pronóstica global del modelo de 4 estratos basado en el registro COMPERA 2.0. HAPh, hipertensión arterial pulmonar heredable; HAPi, hipertensión arterial pulmonar idiopática; NGS, secuenciación de nueva generación; PP, variante probablemente patógena; P, variante patógena; HAP, hipertensión arterial pulmonar; HP, hipertensión pulmonar; EVOP, enfermedad venooclusiva pulmonar; VSD, variante de significado desconocido; WES, secuenciación de exoma completo.
Un análisis previo tras la introducción de las pruebas genéticas en los pacientes del registro REHAP permitió identificar varios casos asociados a variantes genéticas en los genes BMPR2, KCNK3 y TBX4, que se asociaban a un perfil de supervivencia diferente17. Además, la identificación del gen EIF2AK4 como causa de la EVOP heredable fue uno de los hallazgos más notables en el campo de la genética en la HAP en ese momento4. De forma relevante, se describió una prevalencia relativamente elevada de la EVOP heredable en la población del registro REHAP, sobre todo en los pacientes de raza romaní. El descubrimiento de una variante homocigota muy conservada en esta población extremadamente endogámica sugirió un posible efecto fundador18. El presente estudio constituye una continuación de estos esfuerzos por asociar el fenotipo y genotipo en esta población. En la actualidad, es más frecuente realizar pruebas genéticas con técnicas de gran precisión, que han aumentado el rendimiento diagnóstico genético. Y lo que es más importante, el número de genes identificados en estos pacientes se ha multiplicado, y ello nos ha permitido reclasificar varios casos en función de los resultados genéticos (figura 2; ).
En estudios previos se ha demostrado una prevalencia significativa de variantes genéticas en pacientes con HAP asociada a fenfluramina19. En este sentido, algunos autores han propuesto un posible mecanismo genético en pacientes con HAP asociada a la exposición a metanfetaminas20. En consecuencia, las recomendaciones actuales sugieren el uso de estudios genéticos en pacientes con una HAP relacionada con una exposición previa a tóxicos o medicamentos. En nuestro estudio no detectamos ninguna variante genética en este grupo. Por consiguiente, la genética no influyó en la clasificación clínica de estos pacientes. No obstante, es importante señalar que todos los pacientes de este grupo habían tenido una exposición previa a aceite de colza desnaturalizado, y constituyen la cohorte más grande estudiada hasta el momento de HAP asociada al síndrome de aceite tóxico.
En comparación con otros registros previos que han estudiado el riesgo existente en la la primera valoración, como el COMPERA de 4 estratos9, el REVEAL Lite 2.010 o el modelo simplificado basado en el FPHR21, la aplicación del modelo de 4 estratos en la población evaluada genéticamente del REHAP mostró una bondad de ajuste similar evaluada mediante el estadístico C de Harrel. Este índice C fue comparable a los de otros registros. No obstante, la identificación de los pacientes de riesgo intermedio-bajo y de riesgo intermedio-alto fue deficiente con el empleo del modelo original (p=0,635). En cambio, hubo una tendencia a una mejor discriminación de los pacientes de riesgo intermedio-bajo y de riesgo intermedio-alto con el empleo del modelo ampliado que incluía la genética (p=0,053). No se observaron diferencias estadísticas entre los estratos de riesgo intermedio-alto y de riesgo alto con este último modelo (figura 5). Teniendo en cuenta la identificación de las variantes genéticas en la HAP y su posible asociación con el pronóstico, un metanálisis previo con la inclusión de aproximadamente 700 pacientes puso de manifiesto que la presencia de variantes P o PP en el gen que codifica el BMPR2 se asoció a un peor pronóstico6. Se consideró que la gravedad hemodinámica desempeñaba un papel clave en la mayor mortalidad observada en los pacientes con variantes en el gen BMPR2, sobre todo en los pacientes de menor edad. Aunque la repercusión pronóstica de otros genes no se ha evaluado aún, ha habido publicaciones sobre la influencia de las variantes genéticas de otros genes en la gravedad hemodinámica3. En nuestro estudio, la presión arterial pulmonar media fue ligeramente mayor y el índice cardíaco fue ligeramente inferior en los pacientes con variantes genéticas P o PP. Además, estas diferencias hemodinámicas no parecieron ser clínicamente trascendentes, por lo que podría haber otros posibles mecanismos que se asociaran a una mejor estratificación del riesgo con el empleo de la genética. Una posible explicación de esta observación es que la edad de los pacientes incluidos en nuestro estudio fue significativamente inferior a la de los pacientes incluidos en el registro COMPERA 2.0 (65,7 años)9 y la de los incluidos en el registro FPHR, que se utilizó para la validación externa del modelo de 4 estratos (61±15 años)15. De hecho, nuestra población se parece a la incluida en el cluster 1 del registro COMPERA, una población sin comorbilidades y caracterizada por su gravedad hemodinámica22. Es posible que en los pacientes del cluster 1 pudieran ser necesarios otros factores adicionales para estratificar mejor a los pacientes de riesgo intermedio. La huella genética podría haber influido en la estratificación de los pacientes de riesgo intermedio, ya que los casos heredables tienen generalmente un mayor tiempo de evolución de la enfermedad y requieren con más frecuencia trasplante pulmonar. Además, la introducción de los pacientes con EVOP heredable podría haber estado relacionada con la capacidad pronóstica comparativa de la genética en los pacientes de menor edad. No obstante, se incluyó a un número similar de pacientes con EVOP esporádica y con EVOP familiar, y los primeros presentaban una edad significativamente mayor, tenían una clase funcional peor y presentaban concentraciones más altas de biomarcadores cardíacos, aspectos estos que se han asociado también a una peor supervivencia.
Otro aspecto sumamente interesante de nuestro estudio es la inclusión de la EVOP. Un total de 35 pacientes se clasificaron clínicamente como pacientes con EVOP después del estudio diagnóstico inicial. Catorce de ellos fueron reclasificados como casos de enfermedad heredable después de la identificación de una variante homocigota en el gen EIF2AK4 (40,0%). En comparación con los datos previos de Eyries et al.4, el rendimiento diagnóstico del análisis genético en los pacientes con una EVOP esporádica fue ligeramente superior en nuestro estudio (en comparación con el 25,0% de su estudio). Así pues, es importante señalar que en su estudio todos los casos tuvieron confirmación histológica. En nuestra serie, de los 20 pacientes con una EVOP heredable (confirmada genéticamente), 9 precisaron trasplante bipulmonar (45,0%) y 5 fallecieron durante el seguimiento (25,0%). En cambio, 5 de los 22 pacientes del grupo con EVOP esporádica precisaron trasplante pulmonar (22,7%) y 8 fallecieron (36,4%). En todos los pacientes trasplantados y en 5 de los 8 supervivientes con una EVOP esporádica, se dispuso de confirmación histológica. Aunque estos pacientes podrían asociarse a un fenotipo específico, que incluye la aparición de edema pulmonar después del inicio del tratamiento con vasodilatadores pulmonares, este trastorno sumamente infrecuente suele clasificarse como HAP idiopática23. Como pone de manifiesto nuestro estudio, la reclasificación de los pacientes del grupo de HAP inicial al de EVOP heredable es relativamente frecuente. Aquí incluimos un número sustancial de pacientes con EVOP esporádica, que tenían unas características similares a las de los pacientes con EVOP heredable, pero diferían en algunas otras características importantes. Así, los pacientes con EVOP esporádica eran de mayor edad y tenían una concentración de ntproBNP notablemente superior en el momento del diagnóstico, tal como han descrito Montani et al.24 (mediana de edad de 35,0 años en nuestro estudio frente a 26,0 en el suyo, incluidos pacientes pediátricos en la EVOP heredable; 55,0 años frente a 60,0 años en la EVOP esporádica). Teniendo esto en cuenta, la inclusión de la EVOP en nuestra población parece pertinente, no solo para determinar si la presencia de un resultado genético definitivo podría asociarse a un peor resultado en los pacientes clasificados inicialmente como casos de HAP idiopática, sino también en los pacientes con una sospecha clínica inicial de EVOP. Esta es la primera vez que se ha incluido a pacientes con una EVOP en un modelo multivariante validado para los pacientes con HAP. Con objeto de determinar el valor de estos modelos en la EVOP, los aplicamos específicamente a la población con EVOP, y mostramos que el modelo de 4 estratos de COMPERA 2.0 podía ser útil también en esta población. En el futuro deberán crearse modelos específicos en los pacientes con un diagnóstico de EVOP para verificar los resultados.
El modelo de 4 estratos fue introducido recientemente en la guía sobre la hipertensión pulmonar de 2022 de la Sociedad Europea de Cardiología y la European Respiratory Society como herramienta preferente para la estratificación del pronóstico durante el seguimiento25. Hipotéticamente, la adición de la genética, como variable estática no invasiva que generalmente se determina después del diagnóstico inicial, podría permitirnos estratificar mejor a los pacientes durante el seguimiento. El estudio no se diseñó para detectar diferencias entre diferentes grupos de riesgo en los pacientes con HAP o con EVOP. No obstante, a la vista de lo observado en nuestro trabajo, una muestra más grande podría haber hallado diferencias entre los estratos de riesgo en la HAP y en la EVOP.
LimitacionesHay algunas limitaciones que podrían limitar los resultados de nuestro trabajo. En primer lugar, dado que incluimos exclusivamente a pacientes en los que se habían realizado pruebas genéticas, un posible sesgo de selección podría haber favorecido la inclusión de pacientes jóvenes con HAP y EVOP heredables. En segundo lugar, incluimos un número limitado de pacientes del registro, debido a la presencia de varios valores no disponibles de marcadores cardiacos y al número limitado de pacientes en los que se realizaron pruebas de detección sistemática genéticas. No obstante, el porcentaje de inclusión fue similar al observado en el estudio de los investigadores del COMPERA 2.0 que introdujeron el modelo de 4 estratos (142 pacientes de entre 904 posibles candidatos en nuestro estudio; 1655 de una cohorte inicial de 10.825 pacientes en el registro COMPERA)9. Por último, de manera deliberada no incluimos variables pronósticas como la función ventricular derecha o las pruebas de esfuerzo cardiorrespiratorias, puesto que el objetivo principal del estudio fue comparar el valor pronóstico de los modelos simplificados validados con anterioridad con el de nuestro modelo que incluyó las pruebas genéticas en la situación inicial.
CONCLUSIONESEste estudio aporta nuevas hipótesis sobre la utilidad de las pruebas genéticas para la reclasificación clínica y el pronóstico. A pesar de que la adición de los resultados genéticos no mejoró la utilidad del modelo actual de 4 estratos, este estudio sugiere que el uso de la genética podría conducir a una estratificación más exacta del riesgo intermedio, no solo en el subgrupo de pacientes de menor edad con una HAP idiopática, heredable o inducida por fármacos, sino también en los pacientes con una EVOP heredable o esporádica.
FINANCIACIÓNEste proyecto fue financiado por los proyectos «Bases Genético-Moleculares de la Medicina de Precisión en la Hipertensión Arterial Pulmonar» y «Hacia una clasificación ómica de la hipertensión arterial pulmonar», Instituto de Salud Carlos III, Ministerio de Ciencia e Innovación, Gobierno de España. Número de las subvenciones: PI 18/01233, PI21-01593, y PI21-01690. A. Cruz-Utrilla tiene un contrato de investigación-formación «Rio Hortega» (CM20/00164) del Ministerio de Ciencia e Innovación de España (Instituto de Salud Carlos III).
CONTRIBUCIÓN DE LOS AUTORESJ. Antonio Tenorio-Castaño y P. Escribano-Subias contribuyeron por igual a la realización de este artículo. Contribuciones sustanciales al concepto o diseño del trabajo; o en la obtención, análisis o interpretación de los datos para el estudio: A. Cruz-Utrilla, N. Gallego-Zazo, C. Pérez-Olivares, I. Hernández-González, P. Bedate, A. Martínez Meñaca, M. López Meseguer, P. Lapunzina, M. Pérez Núñez, N. Ochoa Parra, J. Tenorio-Castaño, P. Escribano-Subias. Redacción de la versión preliminar del trabajo o revisión respecto a contenido intelectual importante: A. Cruz-Utrilla, N. Gallego-Zazo, J. Tenorio-Castaño, P. Escribano-Subias. Aprobación final de la versión a publicar: A. Cruz-Utrilla, N. Gallego-Zazo, C. Pérez-Olivares, I. Hernández-González, P. Bedate, A. Martínez Meñaca, M. López Meseguer, P. Lapunzina, M. Pérez Núñez, N. Ochoa Parra, J. Tenorio-Castaño, P. Escribano-Subias. Aceptación de la responsabilidad respecto a todos los aspectos del trabajo por lo que respecta a garantizar que las cuestiones relativas a la exactitud o integridad de cualquier parte del mismo se hayan investigado y resuelto adecuadamente. A. Cruz-Utrilla, N. Gallego-Zazo, C. Pérez-Olivares, I. Hernández-González, P. Bedate, A. Martínez Meñaca, M. López Meseguer, P. Lapunzina, M. Pérez Núñez, N. Ochoa Parra, J. Tenorio-Castaño, P. Escribano-Subias.
CONFLICTOS DE INTERESESA. Cruz-Utrilla ha recibido financiación de MSD, Janssen, Gossamer Bio y Ferrer. N. Gallego Zazo no recibió financiación. C. Pérez-Olivares ha recibido financiación de Janssen y MSD. I. Hernández-González no recibió financiación. P. Bedate no recibió financiación. A. Martínez Meñaca ha recibido financiación de MSD, Janssen, Gossamer Bio, AOP Orphan, Chiesi, Rovi y Leo Pharma. M. López Meseguer ha recibido financiación de MSD, Janssen y Ferrer. P. Lapunzina no recibió financiación. M. Pérez Núñez no recibió financiación. N. Ochoa Parra no recibió financiación. D. Valverde no recibió financiación. J. Tenorio-Castaño no recibió financiación. Jair Tenorio recibió una subvención de FEDER y de FCHP. P. Escribano-Subias ha recibido financiación de MSD, Janssen, Gossamer Bio, AOP y Ferrer.
Este proyecto contó con el apoyo del registro de hipertensión arterial pulmonar de España (REHAP), y de los Centros de Investigación Biomédica en Red de Enfermedades Cardiovasculares (CIBERCV), Enfermedades Raras (CIBERER) y Enfermedades Respiratorias (CIBERES), todos ellos vinculados al Instituto de Salud Carlos III (Ministerio de Ciencia e Innovación de España). Además, quisiéramos dar las gracias a nuestros pacientes por hacer posible el estudio.