El mecanismo desencadenante de la angina inestable es la interrupción transitoria de la perfusión miocárdica por un trombo superpuesto a una placa aterosclerótica coronaria fisurada. Recientemente se ha identificado un proceso inflamatorio que precede a la rotura endotelial y que puede desempeñar un papel desencadenante y/o perpetuador de los fenómenos descritos, alterando la adhesividad del endotelio para los leucocitos o plaquetas, y estimulando la actividad procoagulante y vasoconstrictora del endotelio.La existencia de este mecanismo inflamatorio se sustenta en evidencias histológicas, hematológicas, y humorales, entre ellas, la existencia de niveles altos de proteína C reactiva (PCR), una de las proteínas plasmáticas cuya concentración aumenta más del 25% durante el fenómeno inflamatorio.Varios estudios de angina inestable han demostrado el aumento de la concentración sérica de marcadores de inflamación como PCR y amiloide A, así como una asociación entre el nivel de PCR basal y el pronóstico ulterior. Como resumen puede mencionarse que el aumento del valor de PCR es un predictor independiente de evolución adversa en pacientes con angina inestable o infarto sin onda Q. Además, la persistencia del aumento del nivel de PCR podría tener mayor valor pronóstico que el aumento transitorio. Se ha observado valor pronóstico aditivo entre PCR y troponinas.Aunque la valoración clínica es fundamental para la estimación del riesgo, los marcadores séricos como la PCR pueden ser útiles para complementar la información de las pruebas convencionales.
Palabras clave
inflamación
Angina inestable
Pronóstico
INFLAMACIÓN EN LA ANGINA INESTABLE
El mecanismo desencadenante de la angina inestable es la interrupción transitoria de la perfusión miocárdica por un trombo superpuesto a una placa aterosclerótica coronaria fisurada o erosionada 1,2. A su vez, el trombo se forma debido a que el endotelio que cubre el centro lipídico de la placa se fragmenta, permitiendo el contacto de la sangre circulante con factores vasoconstrictores y protrombóticos existentes en la superficie denudada 3,4. Normalmente estos agentes no influyen en el balance trombótico y de vasomotilidad debido a la presencia de un endotelio indemne 5-8. Recientemente se ha identificado un proceso inflamatorio que precede a la rotura endotelial y que puede desempeñar un papel desencadenante y/o perpetuador de los fenómenos descritos 9-13.
Evidencias a favor de la inflamación en los síndromes isquémicos
Aunque la arteriosclerosis ha sido considerada una enfermedad por acumulación de lípidos, existen ahora datos claros que indican que la inflamación está presente desde su comienzo (la placa grasa inicial es una lesión inflamatoria con infiltrado de monocitos y linfocitos T).
La inflamación es una respuesta inespecífica que puede ser evocada por múltiples factores aislados o en combinación. Entre ellos se encuentran las partículas LDL aumentadas y modificadas, una causa de daño endotelial 14,15 cuyo efecto comienza al incorporarse a la pared vascular arterial, y prosigue con su oxidación y captación por los macrófagos y la formación de células esponjosas 15. Ésta es la primera etapa de la arteriosclerosis y constituye, en sí misma, una respuesta inflamatoria que luego se amplifica y genera mayor reclutamiento de monocitos. Como parte de este mecanismo, se produce una modificación de las lipoproteínas por mediadores de la inflamación como el factor de necrosis a, la interleucina 1 y el factor estimulan-te de colonias de macrófagos 16,17. Las lipoproteínas modificadas aumentan su adhesión al endotelio y al músculo liso, incrementan la transcripción del gen receptor-LDL y producen expresión de urocinasa y citocinas inflamatorias como la interleucina 1 18. Este ciclo de inflamación-modificación de lipoproteínas-mayor inflamación se perpetúa por la presencia de estos lípidos en la pared arterial.
Aunque la hipótesis lipídica es la más difundida, otros elementos son capaces de inflamar la pared arterial como los radicales libres, la hipertensión arterial 19, la diabetes, alteraciones genéticas o la elevación de la homocisteína en plasma. Hay también evidencia serológica indicativa de que infecciones con microorga-nismos como Helycobacter pylori, Chlamydia pneumoniae, virus del herpes o citomegalovirus 20-22 son comunes en pacientes con aterosclerosis. Cualquiera de estos agentes puede alterar la adhesividad del endotelio para los leucocitos o plaquetas y estimular la actividad procoagulante y vasoconstrictora del endotelio. Si la respuesta inflamatoria inicial no extrae el tóxico actuante, el proceso se autoperpetúa y estimula la proliferación muscular en la pared arterial, y su consecuente transformación en una placa aterosclerótica consolidada.
La existencia de este mecanismo inflamatorio se sustenta en evidencias de distinto tipo. En la autopsia de lesiones coronarias se han observado signos de inflamación activa en las lesiones arterioscleróticas, además de infiltración inflamatoria en la adventicia coronaria 9. Moreno et al 10 observaron mayor frecuencia de áreas ricas en macrófagos en las lesiones coronarias de pacientes con infarto sin onda Q, o angina inestable. Van der Wal 11, en pacientes fallecidos por infarto agudo de miocardio, observaron que en el sitio del accidente endotelial los macrófagos y los linfocitos T eran la especie celular más frecuente. En el material obtenido de la aterectomía direccional, el número de mastocitos y células T, así como el contenido de factor de necrosis tumoral alfa (TNF a) en los mastocitos y de metaloproteinasa en los macrófagos, era mayor en los pacientes refractarios e inestables 12. Se cree que estos factores contribuyen a la degradación de la matriz extracelular y a la precipitación de una rotura endotelial 13.
Existen también evidencias hematológicas. En pacientes con síndromes isquémicos agudos, se ha comprobado la presencia en la circulación de monocitos, linfocitos y macrófagos activados 23,24. Biasucci et al 25 demostraron que en pacientes con angina inestable o infarto la reducción del contenido de mieloperoxidasa, (un índice del nivel de activación de los neutrófilos) era mayor que en pacientes con angina variante o crónica. Los monocitos, precursores de los macrófagos, están involucrados en la evolución de la aterosclerosis y son potentes activadores de la coagulación a través de su capacidad para generar factor tisular. Jude et al 26 encontraron que existe activación de los monocitos circulantes en la angina inestable, pero no en el infarto agudo o en la angina crónica. Leatham et al 27 demostraron que la proporción de monocitos con expresión de factor tisular era mayor en pacientes con angina inestable o infarto, comparados con controles con angina crónica o normales. El hallazgo de estas células inflamatorias circulantes con mayor contenido de un potente factor protrombótico, como el factor tisular, provee un nexo entre la etapa inflamatoria y las complicaciones trombóticas.
Por último, existen también evidencias humorales, entre las que se destaca la existencia de niveles altos de PCR en pacientes con síndromes isquémicos agudos.
SIGNIFICADO BIOLÓGICO DE LA PROTEÍNA C REACTIVA
La inflamación es un fenómeno complejo en el que se han descrito una gran cantidad de alteraciones sistémicas asociadas. A pesar de que estos cambios pueden observarse tanto en la inflamación aguda como en la crónica, se los conoce en general como respuesta de fase aguda. Entre los cambios de fase aguda más importantes se encuentran las alteraciones en la concentración de un grupo de proteínas séricas denominadas reactantes de fase aguda 28. Éstas se definen como aquellas proteínas plasmáticas cuya concentración aumenta por lo menos un 25% durante el fenómeno inflamatorio. Una de ellas, la proteína C reactiva (PCR), puede aumentar hasta 1.000 veces su concentración. La PCR y otras proteínas de fase aguda se originan en el hepatocito, secundariamente al estímulo de citocinas como interleucina 6, interleucina 1 b, factor de necrosis tumoral, interferón-g, etc. Las citocinas poseen un complejo sistema de interacción entre sí y con diversos sistemas, y participan en el desarrollo de las manifestaciones más visibles y conocidas de la inflamación, como la fiebre, la somnolencia, la letargia o la anorexia. La función de las proteínas de fase aguda es poco conocida. Se sabe que algunas pueden tener efectos proinflamatorios, en tanto que otras inhiben es-te proceso. La PCR, por ejemplo, posee la propiedad de ligarse a la fosfocolina y reconocer patógenos externos 29. También posee otros efectos como inhibir la adhesión de los leucocitos a la pared endotelial, inhibir la generación de superóxidos en los neutrófilos, y estimular en los monocitos la síntesis de citocinas inflamatorias y factor tisular 30. Algunas de estas propiedades son probablemente importantes para inhibir el proceso inflamatorio.
El aumento de la velocidad de sedimentación de los eritrocitos es un indicador de inflamación muy conocido, cuyo nivel depende fundamentalmente de la concentración plasmática de un reactante de fase aguda, el fibrinógeno. También influyen en su valor las características de los eritrocitos y la edad. En cambio, la PCR no está sujeta a estas influencias, por lo que se ha transformado en un parámetro más fiable para la estimación o el seguimiento de la inflamación. Procesos inflamatorios menores como los traumas mínimos son capaces de elevar los valores de PCR, revelando la alta sensibilidad de este marcador. Otra de las proteínas de fase aguda, el amiloide A, experimenta cambios muy similares a los de la PCR o el fibrinógeno.
El nivel de las citocinas plasmáticas también ha sido estudiado con este propósito, aunque su medición es más dificultosa por su corta vida media. Biasucci et al 31 comunicaron el hallazgo de niveles elevados de interleucina 6 en pacientes con angina inestable y enzimas miocárdicas normales. Basándose en estos datos los autores concluyen que la existencia de un mecanismo inflamatorio en la angina inestable no se debe a la presencia de necrosis miocárdica. El daño por reperfusión consecutivo a la isquemia prolongada y severa, y a la activación de la coagulación, puede generar un aumento de las citocinas y de los niveles de los reactantes de fase aguda 32-34. Sin embargo, los estudios de Biasucci et al 35 y Liuzzo et al 36 indican que ninguno de estos mecanismos es un desencadenante de inflamación en pacientes con angina inestable.
El significado pronóstico de los marcadores de inflamación en la angina inestable
El aumento de la concentración sérica de marcadores de inflamación se ha observado en varios estudios de angina inestable. Berck et al 37 fueron de los primeros en señalar la existencia de niveles anormales de PCR en la angina inestable. Liuzzo et al 38 midieron el nivel de dos reactantes de fase aguda, la PCR y el amiloide A, en 31 pacientes con angina inestable y niveles normales de troponina T (TnT) y CPK MB en el momento del ingreso. Como comparación analizaron también un grupo con angina crónica estable y otro con infarto agudo de miocardio. En el ingreso, la PCR estaba elevada en 65% de los pacientes con angina inestable, en el 13% de las anginas estables y en el 76% de los pacientes con infarto. El aumento de la PCR no se correlacionó con la severidad angiográfica de la coronariopatía. La conclusión de los autores fue que la angina inestable se asocia a un mecanismo inflamatorio no relacionado con la presencia de necrosis ni con la severidad o extensión de las lesiones coronarias. Además, este estudio fue uno de los primeros en indicar el valor pronóstico de la PCR elevada, ya que entre los pacientes con PCR anormal se observaron más episodios isquémicos en el hospital.
En el mismo estudio se encontró elevación de la PCR en todos los pacientes con infarto precedido por angina inestable y en sólo el 53% de los pacientes sin angina inestable previa. La elevación de la PCR fue mucho más frecuente en los pacientes que presentaron angina inestable en los 7 días anteriores (el 100 frente al 55%; p = 0,002). Estos datos fueron corroborados en un estudio posterior de 36 pacientes con infarto agudo de miocardio admitidos con CPK y TnT normales y menos de 3 h de evolución, por lo que los autores sugieren que el aumento de la respuesta de fase aguda en los pacientes con angina preinfarto implica una diferencia en la patogenia 39.
La asociación entre el nivel de PCR basal y el pronóstico ulterior fue observada también en el estudio de Haverkate 40. En 1.030 pacientes con angina inestable admitidos con nivel de PCR en el quintilo superior, se observó peor evolución durante un seguimiento de 24 meses. En un subestudio del ensayo TIMI 11B, Morrow et al 41 examinaron el significado pronóstico de la elevación de la PCR en el momento del ingreso hospitalario de 437 pacientes con angina inestable o infarto agudo de miocardio (IAM) no Q, y observaron que la mortalidad a los 14 días fue mayor en pacientes con PCR por encima de 1,5 mg/l (el 5,6 frente al 0,3%; p = 0,001). Lo más notable es que la misma diferencia se observó en el grupo con TnT negativa. Al combinar los valores de TnT y PCR, se encontró que el subgrupo con mayor mortalidad fue aquel en el que ambos marcadores estaban elevados. Posteriormente resultados similares fueron comunicados por estos autores para el amiloide A.
En el estudio FRISC 42, se analizaron los valores plasmáticos de PCR y fibrinógeno en 965 pacientes con angina inestable o IAM sin onda Q, con el último episodio de dolor en las 72 h previas. Se demostró una asociación significativa entre el nivel inicial de PCR y la mortalidad a los 5 meses, pero no se observó asociación con la incidencia de infarto en el seguimiento.
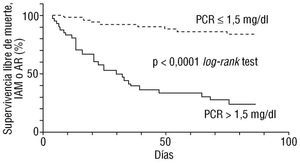
En 211 pacientes con angina inestable, Verheggen et al 43 midieron el nivel basal de marcadores de inflamación, activación de la coagulación y función endotelial, y los correlacionaron con la evolución clínica posterior intrahospitalaria. Encontraron que el riesgo de presentar angina refractaria era mayor en pacientes con elevación de la PCR u otros marcadores de inflamación, pero no en aquéllos con elevación de los indicadores de activación de coagulación o de función endotelial. Estos hallazgos se confirmaron en el subgrupo con TnT normal e indican que la actividad inflamatoria puede ser mejor reflejo del proceso subya-cente y de la evolución posterior, aun en ausencia de necrosis miocárdica. En un estudio reciente de nuestro centro 44, se incluyeron prospectivamente 189 pacientes con angina inestable para analizar el valor pronóstico de la PCR y compararlo con el de los predictores clínicos habituales. El nivel de PCR en la admisión no se relacionó con la evolución en el hospital, pero en el análisis multivariado resultó un marcador pronóstico a los 90 días independiente de los predictores habituales. Otro hallazgo interesante fue el fuerte significado pronóstico adverso de la persistencia de la PCR elevada (> 1,5 mg/l) medida en el momento del alta (fig. 1).

Fig. 1. Curvas de Kaplan-Meier. Supervivencia libre de eventos en pacientes con angina inestable y proteína C reactiva (PCR) mayor o menor de 1,5 mg/l en el momento del alta. IAM: infarto agudo de miocardio; AR: angina refractaria.
Observaciones similares surgieron en otro estudio de Biasucci et al 45. Entre los pacientes dados de alta con PCR mayor de 3 mg/l, un 69% fueron reingresados por un nuevo episodio isquémico frente a sólo un 15% de los que fueron dados de alta con PCR normal.
En otro ensayo de nuestro centro 46, el valor pronóstico de la medición de la PCR en el momento del alta del hospital se comparó con el de la isquemia inducida por ejercicio o dobutamina. La medición de la PCR demostró valor similar o superior al de los tests evocadores de isquemia.
El valor predictivo de la PCR también se comprobó después de la angioplastia 47. En un subgrupo de 447 pacientes tratados con placebo en el ensayo CAPTURE (angioplastia y abciximab en la angina refractaria), el nivel de PCR preprocedimiento fue predictor independiente de los eventos tardíos (6 meses), en presencia de TnT elevada o normal.
Hallazgos similares a los observados con la PCR se han comunicado con otros marcadores de inflamación. La elevación de la interleucina 6, la citocina causante de la producción de PCR, se asoció a peor evolución en un estudio de Biasucci et al 31. En un subestudio del ensayo ESSENCE, Montalescot et al 48 observaron en 68 pacientes un brusco aumento del nivel de PCR y del nivel del factor de Von Willebrand. Los pacientes con elevación de estos marcadores tuvieron peor evolución a los 14 y 30 días. Estos datos indican un mismo papel pronóstico para los diferentes marcadores de inflamación.
Debe tenerse en cuenta que el aumento de nivel de la PCR también puede expresar inflamación secundaria a necrosis miocárdica. Se ha observado que la PCR comienza a elevarse varias horas después del comienzo del infarto de miocardio 49. En los estudios de PCR en la angina inestable, los pacientes seleccionados difieren tanto en lo referente a la presencia de necrosis miocárdica como en cuanto al tiempo de evolución transcurrido desde los síntomas que motivaron la inclusión en el estudio. Sabemos de los estudios de infarto que la elevación tardía (12 h) de la PCR tiende a asociarse a complicaciones de la necrosis, como rotura o disfunción ventricular, pero no a la recurrencia de isquemia. Por tanto, los distintos mecanismos causantes de la elevación de la PCR pueden explicar las diferencias en el valor predictivo observado en los diversos estudios.
RESUMEN
La información analizada puede sintetizarse del modo siguiente:
1. El aumento del nivel de PCR es un predictor independiente de evolución adversa en pacientes con angina inestable o infarto sin onda Q.
2. La magnitud de la elevación es importante, y por tanto, aunque no existe un punto de corte uniforme, las mediciones de PCR deben ser cuantitativas.
3. Después de un episodio de angina inestable, la persistencia del aumento del nivel de la PCR parece tener mayor valor pronóstico que el aumento aislado en el momento del ingreso.
4. Existe valor pronóstico aditivo entre la PCR y las troponinas.
5. En el infarto, el aumento tardío de la PCR (más de 12 h) es indicativo de necrosis y predice complicaciones mecánicas, en tanto que el aumento basal o precoz indica la existencia de inflamación coronaria y predice recurrencia de isquemia. CONCLUSIÓN
Aunque la valoración clínica es fundamental para la estimación del riesgo 50,51 la precisión de los predictores clínicos puede resultar insuficiente 52. En los cuadros isquémicos agudos el pronóstico depende de la persistencia de la inestabilidad en la placa. Por tanto, los métodos de estratificación de riesgo habituales basados en el estudio de la reserva coronaria 52-55, pueden resultar inadecuados para detectar la inestabilidad de la placa 56,57. Los nuevos marcadores humorales pueden ser de importancia para suplir esta carencia de las pruebas convencionales. Aunque algunos autores ya han comunicado el valor de la combinación de estos marcadores entre sí y con otros predictores conocidos 58-62, sus ventajas relativas y el valor complementario de las troponinas y la PCR merecen establecerse con nuevos estudios prospectivos.
Bibliografía
[1]
Fuster V, Badimon L, Cohen M, Ambrose JA, Badimon JJ, Chesebro JH..
Insights into the pathogenesis of acute ischemic syndromes..
Circulation, (1988), 77 pp. 1213-1220
[2]
Fuster V, Badimon L, Badimon JJ, Chesebro JH..
The pathogenesis of coronary artery disease and the acute coronary syndromes..
N Engl J Med, (1992), 326 pp. 242-250
[3]
Constantinides P..
Plaque fissures in human coronary thrombosis..
J Atheroscler Res, (1966), 6 pp. 1-17
[4]
Davies MJ, Thomas AC..
Plaque fissuring-the cause of acute myocardial infarction, sudden ischemic death, and crescendo angina..
Br Heart J, (1985), 53 pp. 363-373
[5]
Davies MJ, Thomas AC..
Thrombosis and acute coronary artery lesion in sudden cardiac ischemic death..
N Engl J Med, (1984), 310 pp. 1137-1140
[6]
Chesebro JH, Zoldhelyi P, Fuster V..
Plaque disruption and thrombosis in unstable angina pectoris..
Am J Cardiol, (1991), 68 pp. 9C-15C
[7]
Davies MJ, Thomas AC, Knapman PA, Hangartner JR..
Intramyocardial platelet aggregation in patients with unstable angina suffering sudden ischemic cardiac death..
Circulation, (1986), 73 pp. 418-427
[8]
Falk E..
Unstable angina with fatal outcome: dynamic coronary thrombosis leading to infarction and/or sudden death..
Circulation, (1985), 71 pp. 699-708
[9]
Kohchi K, Takebayashi S, Hiroki T, Nobuyoshi M..
Significance of adventitial inflammation of the coronary artery in patients with unstable angina: results at autopsy..
Circulation, (1985), 71 pp. 709-716
[10]
Moreno PR, Falk E, Palacios IF, Newell JB, Fuster V, Fallon JT..
Macrophage infiltration in acute coronary syndromes: implications for plaque rupture..
Circulation, (1994), 90 pp. 775-778
[11]
Van der Wa.l, Becker AE, Van der Los Ch, Das PK..
Site of intimal rupture or erosion of thrombosed coronary atherosclerotic plaques is characterized by an inflammatory process irrespective of the dominant plaque morphology..
Circulation, (1994), 89 pp. 36-44
[12]
Kaartinen M, Van der Wal A, Van der Loos C, Piek J, Koch K, Becker A et al..
Mast cell infiltration in acute coronary syndromes: implications for plaque rupture..
J Am Coll Cardiol, (1998), 32 pp. 606-612
[13]
Shah PK, Falk E, Badimon JJ, Fernández-Ortiz A, Mailhac A, Villareal-Levy G et al..
Human monocyte-derived macrophages induce collagen breakdown in fibrous caps of atherosclerotic plaques: potential role of matrix-degrading metalloproteinases and implications for plaque rupture..
Circulation, (1995), 92 pp. 1565-1569
[14]
Navab M, Berliner JA, Watson AD, Hama SY, Territo MC, Lusis AJ et al..
The Yin and Yang of oxidation in the development of the fatty streak: the review based on the 1994 George Lyman Duff Memorial Lecture..
Arterioscler Thromb Vasc Biol, (1996), 16 pp. 831-842
[15]
Griendling KK, Alexander RW..
Oxidative stress and cardiovascular disease..
Circulation, (1997), 96 pp. 3264-3265
[16]
Stopeck AT, Nicholson AC, Mancini FP, Hajjar DP..
Cytokine regulation of low density lipoprotein receptor gene transcription in HepG2 cells..
J Biol Chem, (1993), 268 pp. 17489-1794
[17]
Hajjar DP, Haberland ME..
Lipoprotein trafficking in vascular cells: molecular Trojan horses and cellular saboteurs..
J Biol Chem, (1997), 272 pp. 22975-22978
[18]
Geng YJ, Libby P..
Evidence for apoptosis in advanced human atheroma: colocalization with interleukin-1 beta-converting enzyme..
Am J Pathol, (1995), 147 pp. 251-266
[19]
Lacy F, O'Connor DT, Schmid-Schènbein GW..
Plasma hydrogen peroxide production in hypertensives and normotensive subjects at generic risk of hypertension..
J Hypertens, (1998), 16 pp. 291-303
[20]
Hendrix MG, Salimans MM, Van Boven CP, Bruggeman CA..
High prevalence of latently present cytomegalovirus in arterial walls of patients suffering from grade III atherosclerosis..
Am J Pathol, (1990), 136 pp. 23-28
[21]
Melnick JL, Adam E, Debakey ME..
Cytomegalovirus and atherosclerosis..
Eur Heart J, (1993), 14(SuplK) pp. 30-38
[22]
Gupta S, Leatham EW, Carrington D, Mendall M, Kaski JC, Camm J et al..
Elevated Chlamydia pneumoniae antibodies, cardiovascular events, and azithromycin in male survivors of myocardial infarction..
Circulation, (1997), 96 pp. 404-407
[23]
Neri Serneri G, Abbate R, Gori A, Attanasio M, Martini F, Giusti B et al..
Transient intermittent lymphocyte activation is responsible for the instability of angina..
Circulation, (1992), 86 pp. 790-797
[24]
Mazzone A, De Servi S, Ricevuti G, Mazzucchelli L, Fossati G, Pasotti D et al..
Increased expression of neutrophil and monocyte adhesion molecules in unstable coronary artery disease..
Circulation, (1993), 88 pp. 358-363
[25]
Biasucci LM, D'Onofrio G, Liuzzo G, Zini G, Monaco C, Caligiuri G et al..
Intracellular neutrophil myeloperoxidase is reduced in unstable angina and acute myocardial infarction, but its reduction is not related to ischemia..
J Am Coll Cardiol, (1996), 27611 pp. 1137-1140
[26]
Jude B, Agraou B, McFadden EP, Susen S, Bauters C, Lepelley P et al..
Evidence for time dependent activation of monocytes in the systemic circulation in unstable angina but not in acute myocardial infarction or in stable angina..
Circulation, (1994), 90 pp. 1662-1668
[27]
Leatham E, Bath P, Tooze J, Camm A..
Increased monocyte tissue factor expression in coronary disease..
Br Heart J, (1995), 73 pp. 10-13
[28]
Serum C-reactive protein levels in disease. Ann NY Acad Sci 1982, 389: 406-418.
[29]
Acute phase proteins in rheumatic disease. En: Koopman WJ, editor. Arthritis and allied conditions: a textbook of rheumatology (13.
[30]
Cermak J, Key NS, Bach RR, Balla J, Jacb HS, Vercellotti GM..
C-reactive protein induces human peripheral blood monocytes to synthetize tissue factor..
Blood, (1993), 82 pp. 513-520
[31]
Biasucci LM, Vitelli A, Liuzzo G, Attamura S, Caliguri G, Monaco C et al..
Elevated levels of interleukin-6 in unstable angina..
Circulation, (1996), 94 pp. 874-877
[32]
Fossat C, Fabre D, Alimi Y, Bienvenu J, Aillaud MF, Lenoble M et al..
Leukocyte activation study during occlusive arterial disease of the lower limb: effect of pentoxifylline infusion..
J Cardiovasc Pharmacol, (1995), 25 pp. S96-S100
[33]
Dosquet C, Weill D, WautierJL..
Cytokines and thrombosis..
J Cardiovasc Pharmacol, (1995), 25 pp. S13-S19
[34]
Johnson K, Aarden L, Choi Y, De Groot E, Creasey A..
The proinflammatory cytokine response to coagulation and endotoxin in whole blood..
Blood, (1996), 87 pp. 5051-5060
[35]
Biasucci LM, Luizzo G, Caligiuri G, Van de Greef W, Quaranta G, Monaco C et al..
Episodic activation of the coagulation system in unstable angina does not elicit an acute phase reaction..
Am J Cardiol, (1996), 77 pp. 85-87
[36]
Liuzzo G, Biasucci LM, Rebuzzi AG, Gallimore R, Caligiuri G, Lanza G et al..
Plasma protein acute-phase response in unstable angina is not induced by ischemic injury..
Circulation, (1996), 94 pp. 2373-2380
[37]
Berck BC, Weintraub W, Alexander RW..
Elevation of C reactive protein in «active» coronary disease..
Am J Cardid, (1990), 65 pp. 168-172
[38]
Liuzzo G, Biasucci LM, Gallimore JR, Grillo RL, Rebuzzi AG, Pepys MB et al..
The prognostic value of C-reactive protein and serum amyloid A protein in severe unstable angina..
N Engl J Med, (1994), 331 pp. 417-424
[39]
Liuzzo G, Biasucci LM, Gallimore JR, Caligiuri G, Buffon A, Rebuzzi A et al..
Enhanced inflammatory response in patients with pre-infarction angina..
J Am Coll Cardiol, (1999), 34 pp. 1696-1703
[40]
Haverkate F, Thompson SG, Pyke SD, Gallimore JR, Pepys MB..
Production of C-reactive protein and risk of coronary events in stable and unstable angina: European Concerted Action on Thrombosis and Disabilities Angina Pectoris Study Group..
Lancet, (1997), 349 pp. 462-466
[41]
Morrow DA, Rifai N, Antman AN, Weiner DL, McCabe CH, Cannon CP et al..
C-reactive protein is a potent predictor of mortality independently or in combination with troponin T in acute coronary syndromes: a TIMI 11a substudy..
J Am Coll Cardiol, (1998), 31 pp. 1460-1465
[42]
Toss H, Lindahl B, Siegbahn A, Wallentin L..
For the FRISC study group. Prognostic influence of increased fibrinogen and C-reactive protein levels in unstable coronary artery disease..
Circulation, (1997), 96 pp. 4204-4210
[43]
Verheggen P, Maat M, Manger Cats V, Haverkate F, Zwinderman A, Kluft C et al..
Inflammatory status as a main determinant of outcome in patients with unstable angina, independent of coagulation activation and endothelial cell function..
Eur Heart J, (1999), 20 pp. 567-574
[44]
Ferreirós ER, Boissonnet CP, Pizarro R, García Merletti P, Corrado G, Cagide A et al..
Elevated C-reactive protein at discharge is a strong independent predictor of 90-day outcome in unstable angina..
Circulation, (1999), 100 pp. 1958-1963
[45]
Biasucci LM, Liuzzo G, Grillo RL, Caligiuri G, Rebuzzi AG, Buffon A et al..
Elevated levels of C-reactive protein at discharge in patients with unstable angina predict recurrent instability..
Circulation, (1999), 99 pp. 855-860
[46]
Bazzino O, Ferreiros E, García Merletti P, Boissonet C, Pizarro R, Corrado G et al..
Independent and combined prognostic value of CRP and the stress-test in patients recovering from unstable angina (resumen)..
Circulation, (1999), 100 pp. 1-370
[47]
Heeschen Ch, Hamm Ch, Bruemmer J, Simoons M, for the Capture investigators..
Predictive value of C-reactive protein and troponin T in patients with unstable angina. A comparative analysis..
J Am Coll Cardiol, (2000), 35 pp. 1535-1542
[48]
Montalescot G, Philippe F, Ankri A, Vicaut E, Bearez E, Poulard JE et al, for the French Investigators of the ESSENCE Trial..
Early increase of van Willebrand factor predicts adverse outcome in unstable coronary artery disease..
Circulation, (1998), 98 pp. 294-299
[49]
Control of the acute phase response. Serum C-reactive protein kinetics after acute myocardial infarction. J Clin Invest 1978, 61: 235-242.
[50]
Masseri A, Rebuzzi AG, Cianflone D..
Need for a composite risk stratification of patients with unstable coronary syndromes tailored to clinical practice..
Circulation, (1997), 96 pp. 4143-4142
[51]
Théroux P, Fuster V..
Acute coronary syndromes. Unstable angina and non-Q-wave myocardial infarction..
Circulation, (1998), 97 pp. 1195-1206
[52]
Bazzino O, Tajer C, Paviotti C, Mele E, Trivi M, Piombo A..
Clinical predictors of in-hospital prognosis in unstable angina. ECLA 3..
Am Heart J, (1999), 137 pp. 322-331
[53]
Coplan NL, Wallach ID..
The role of exercise testing for evaluating patients with unstable angina..
Am Heart J, (1992), 124 pp. 252-256
[54]
Butman SM, Olson HG, Gardin JM, Piters KM, Hullet M, Butman L..
Submaximal exercise testing after stabilization of unstable angina pectoris..
J Am Coll Cardiol, (1984), 4 pp. 667-673
[55]
Wilcox I, Freedman SB, Allman KC, Collins FL, Leitch JW, Kelly DT et al..
Prognostic significance of a pre-discharge exercise test in risk stratification after unstable angina pectoris..
J Am Coll Cardiol, (1991), 18 pp. 677-683
[56]
Lin SS, Lauer MS, Marwick TH..
Risk stratification of patients with medically treated unstable angina using exercise echocardiography..
Am J Cardiol, (1998), 82 pp. 720-724
[57]
Noninvasive risk stratification after myocardial infarction: which test is best? Am Heart J 1998; 136: 565-569.
[58]
Krone RJ, Gregory JJ, Freedland KE, Kleiger RE, Wackers FJ, Bodenheimer MM et al..
Limited uselfulness of exercise testing and thallium scintigraphy in evaluation of ambulatory patients several months after recovery from acute coronary event: implications for management of stable coronary disease..
J Am Coll Cardiol, (1994), 24 pp. 1274-1281
[59]
Rebuzzi AG, Quaranta G, Liuzzo G, Caligiuri G, Lanza GA, Gallimore JR et al..
Incremental prognostic value of serum levels of troponin T and C-reactive protein on admission in patients with unstable angina pectoris..
Am J Cardiol, (1998), 82 pp. 715-719
[60]
De Winter RJ, Bholasingh R, Koster RW, Gorgels JPM.C, Scouten Y, Hoek FJ et al..
Independent prognostic value of C-reactive protein and troponin I in patients with unstable angina or non-Q wave myocardial infarction..
Cardiovasc Res, (1999), 42 pp. 240-245
[61]
Homvang L, Lüscher M, Clemmensen P, Thygesen K, Grande P, and TRIM Study Group..
Very early risk stratification using combined ECG and biochemical assessment in patients with unstable coronary artery disease (a thrombin inhibition in myocardial ischemia (TRIM substudy)..
Circulation, (1998), 98 pp. 2004-2009