
La amiloidosis cardiaca (AC) es una enfermedad grave y progresiva, más frecuente de lo que se sospechaba en un inicio, para la que se dispone de tratamientos que pueden modificar su pronóstico. Por lo tanto, su diagnóstico precoz es clave1-3. La AC puede presentarse en un mismo paciente junto con otra miocardiopatía más frecuente y permanecer oculta durante años, lo que compromete el pronóstico4. El conocimiento de las red flags (RF) (alertas) de la AC nos puede permitir un diagnóstico más precoz1,2. Se presentan 3 casos en los que la AC no se consideró de inicio.
El primer caso es un varón de 73 años con enfermedad coronaria de tronco y 3 vasos revascularizada mediante cirugía. Se detectó hipertrofia del ventrículo izquierdo (HVI) de 20mm en el ecocardiograma, por lo que se completó el estudio con una cardiorresonancia que confirmó HVI y mostró realce tardío de gadolinio (RTG) mesocárdico septal anterior. El paciente no tenía antecedentes familiares de miocardiopatía hipertrófica (MCH). Se realizó estudio genético mediante next-generation sequencing (NGS) de genes sarcoméricos y fenocopias, que detectó una variante patogénica en TNNC1 (p.Ala8Val), lo que confirmó el diagnóstico de MCH. En el estudio familiar no se identificó a más familiares afectos. Durante el seguimiento, aparecieron datos de insuficiencia cardiaca con patrón de strain típico de AC y «signo de Popeye». Se había intervenido recientemente de estenosis de canal lumbar. Una gammagrafía con 99mTc-DPD con captación de grado 3, una analítica que descartó un componente monoclonal y el estudio genético negativo confirmaron el diagnóstico de AC por transtirretina wild type (ATTR-wt). El paciente falleció a los 79 años en el contexto de una infección respiratoria.
El segundo caso es un varón de 62 años hipertenso, con una hipertrofia septal de 15mm atribuida durante años a su hipertensión. A los 74 años contrajo disnea de esfuerzo. En el ecocardiograma se detectó un grosor de 23mm, movimiento sistólico anterior y gradiente subaórtico de 67mmHg con Valsalva. Se lo remitió a la Unidad de Cardiopatías Familiares, donde se le realizó un estudio genético mediante NGS (genes sarcoméricos y fenocopias) que detectó una variante probablemente patogénica en MYL3 (p.Met173Val), lo que confirmó el diagnóstico de MCH obstructiva. Se inició tratamiento con un bloqueador beta, que controló la clínica de obstrucción. El estudio familiar no detectó a ningún familiar afecto. Dos años más tarde el paciente presentó semiología de insuficiencia cardiaca con fracción aminoterminal del propéptido natriurético cerebral (tipo B) de 3.000 pg/ml, patrón diastólico restrictivo en el ecocardiograma y un «signo de Popeye» en el brazo derecho, que antes no tenía. La cardiorresonancia mostró una cinética alterada del gadolinio y RTG difuso en ambos ventrículos, y la gammagrafía cardiaca con 99mTc-DPD, captación de grado 3. Se descartó el componente monoclonal y se añadió el diagnóstico de AC ATTR-wt. El paciente en la actualidad continúa en seguimiento en consultas.
El tercer caso es una mujer de 75 años hipertensa y con antecedentes de miocardiopatía dilatada familiar por variante genética en titina (p.Trp19433*). Presentaba fibrilación auricular con ventrículo izquierdo no dilatado y fracción de eyección del ventrículo izquierdo (FEVI) < 30%. Es portadora de la variante patogénica familiar. Con tratamiento neurohormonal, la FEVI se recuperó hasta el 50%. A los 78 años sufrió un empeoramiento clínico discordante con la FEVI de ese momento e intolerancia al tratamiento neurohormonal por hipotensión. Además, el electrocardiograma presentaba bajos voltajes con patrón de seudoinfarto y el ecocardiograma, derrame pericárdico ligero, estenosis aórtica moderada y 12mm de septo. Se realizaron una gammagrafía con 99mTc-DPD (informada como captación de grado 1-2) y una analítica, que descartó el componente monoclonal. Se ofreció a la paciente la posibilidad de una confirmación histológica del diagnóstico, que rechazó. La cardiorresonancia evidenció aumento del volumen extracelular y del T1 nativo, RTG difuso y cinética del gadolinio alterada. El estudio genético Sanger del gen TTR fue normal. Con todo ello, se asumió el diagnóstico de ATTR-wt. Actualmente mantiene el seguimiento en consultas, con disnea de pequeños esfuerzos estable.
En la tabla 1 se recogen las características de los pacientes. En la figura 1 se recogen varias pruebas complementarias de los 3 pacientes.
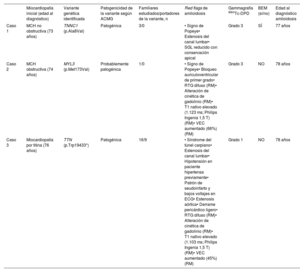
Características de los pacientes
| Miocardiopatía inicial (edad al diagnóstico) | Variante genética identificada | Patogenicidad de la variante según ACMG | Familiares estudiados/portadores de la variante, n | Red flags de amiloidosis | Gammagrafía 99mTc-DPD | BEM (sí/no) | Edad al diagnóstico amiloidosis | |
|---|---|---|---|---|---|---|---|---|
| Caso 1 | MCH no obstructiva (73 años) | TNNC1 (p.Ala8Val) | Patogénica | 3/0 | • Signo de Popeye• Estenosis del canal lumbar• SGL reducido con conservación apical | Grado 3 | SÍ | 77 años |
| Caso 2 | MCH obstructiva (74 años) | MYL3 (p.Met173Val) | Probablemente patogénica | 1/0 | • Signo de Popeye• Bloqueo auriculoventricular de primer grado• RTG difuso (RM)• Alteración de cinética de gadolinio (RM)• T1 nativo elevado (1.123 ms; Philips Ingenia 1,5 T) (RM)• VEC aumentado (66%) (RM) | Grado 3 | NO | 78 años |
| Caso 3 | Miocardiopatía por titina (76 años) | TTN (p.Trp19433*) | Patogénica | 16/9 | • Síndrome del túnel carpiano• Estenosis del canal lumbar• Hipotensión en paciente hipertensa previamente• Patrón de seudoinfarto y bajos voltajes en ECG• Estenosis aórtica• Derrame pericárdico ligero• RTG difuso (RM)• Alteración de cinética de gadolinio (RM)• T1 nativo elevado (1.103 ms; Philips Ingenia 1,5 T) (RM)• VEC aumentado (45%) (RM) | Grado 1 | NO | 78 años |
99mTc-DPD: tecnecio-99m con ácido-3,3-difosfono-1,2-propanodicarboxílico; ACMG: American College of Medical Genetics; BEM: biopsia endomiocárdica; ECG: electrocardiograma; MCH: miocardiopatía hipertrófica; RM: resonancia magnética; RTG: realce tardío de gadolinio; SGL: strain global longitudinal; VEC: volumen extracelular.
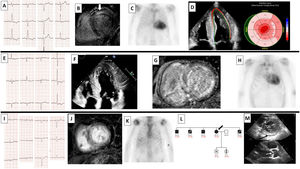
![Caso 1. A: electrocardiograma (ECG). B: cardiorresonancia magnética con realce tardío de gadolinio (RTG) mesocárdico septal anterior (flecha blanca). C: gammagrafía cardiaca con 99mTc-DPD con captación de grado 3. D: strain global longitudinal disminuido con conservación apical. Caso 2. E: ECG. F: ecocardiograma en plano apical de 4 cámaras que muestra engrosamiento concéntrico de ambos ventrículos. G: cardiorresonancia magnética con cinética alterada de gadolinio, RTG difuso y heterogéneo en los ventrículos izquierdo y derecho. H: gammagrafía cardiaca con 99mTc-DPD con captación de grado 3. Caso 3. I: ECG con fibrilación auricular, bajos voltajes y patrón de seudoinfarto. J: cardiorresonancia magnética con RTG difuso y cinética alterada de gadolinio. K: gammagrafía cardiaca con 99mTc-DPD con captación de grado 1-2. L: fragmento del árbol familiar (flecha: paciente 3; en negro, afectados de miocardiopatía por titina; cuadrado negro: varón afecto; círculo negro: mujer afecta; raya diagonal: paciente fallecido; cuadrado blanco: varón sano; N: normal; raya vertical: portador sano; E1 –/+: portador en heterocigosis TTN [p.Trp19433*]; E1 –/–: no portador). M: imagen superior, ecocardiograma con hipertrofia septal de 12mm (asterisco) y derrame pericárdico ligero (flecha); imagen inferior, engrosamiento de la válvula aórtica (flecha). 99mTc-DPD: tecnecio-99m con ácido-3,3-difosfono-1,2-propanodicarboxílico; ECG: electrocardiograma; RM: resonancia magnética; RTG: realce tardío de gadolinio; SGL: strain global longitudinal.](https://static.elsevier.es/multimedia/03008932/0000007700000004/v2_202405080530/S0300893223004529/v2_202405080530/es/main.assets/gr1.jpeg?xkr=eyJpdiI6Ik1HWWtOV1BWbDF6OGJvc20ySXVneUE9PSIsInZhbHVlIjoiaWtxeU1WK2FwT0FLN1dYKzhObGxLb3dxUHY0TmREVlVRZERjeGtoZWU0K1RjL3lLNzhJbHY4Q2M0RENPUTlJZ2lQNHNmeXJGbnBKbDRSTE42RWdEQ2U2ZWFmTGQzUGw1SXA3U2xncG1mSzJjb3Z6TEZmRHFCOGNSU2pwV2ROU2ZncjA2bWlwZnc0amU0QjNrUk10Tkk5REFma1c5clBqRHlCN3Njc3htaWRRTDMySnVoZ3NPbXYwb0szMFBNK0kwUUx3KzNEUGNwMlh6T3NUa0VTSjh6c2JCa0dJc2xRSGZncmpvWTlJNStzM1JKNGU2NDF3VWJpdTAvVy9HMkpjMHJ3WnVERk04V2ZjcnNXblNYTFRHY0V5elhXKyt1TElzcmFveUdyMFNCSlk9IiwibWFjIjoiZjIzZWNiNmUxM2E2Yzc4ZWM4ZWQ4MmI5ODlmMzJmZjg4YjdjMjgzOTk3OGI2MTVhMDgxNzdlNWQxYWNmNzlkNCIsInRhZyI6IiJ9 "Caso 1. A: electrocardiograma (ECG). B: cardiorresonancia magnética con realce tardío de gadolinio (RTG) mesocárdico septal anterior (flecha blanca). C: gammagrafía cardiaca con 99mTc-DPD con captación de grado 3. D: strain global longitudinal disminuido con conservación apical. Caso 2. E: ECG. F: ecocardiograma en plano apical de 4 cámaras que muestra engrosamiento concéntrico de ambos ventrículos. G: cardiorresonancia magnética con cinética alterada de gadolinio, RTG difuso y heterogéneo en los ventrículos izquierdo y derecho. H: gammagrafía cardiaca con 99mTc-DPD con captación de grado 3. Caso 3. I: ECG con fibrilación auricular, bajos voltajes y patrón de seudoinfarto. J: cardiorresonancia magnética con RTG difuso y cinética alterada de gadolinio. K: gammagrafía cardiaca con 99mTc-DPD con captación de grado 1-2. L: fragmento del árbol familiar (flecha: paciente 3; en negro, afectados de miocardiopatía por titina; cuadrado negro: varón afecto; círculo negro: mujer afecta; raya diagonal: paciente fallecido; cuadrado blanco: varón sano; N: normal; raya vertical: portador sano; E1 –/+: portador en heterocigosis TTN [p.Trp19433*]; E1 –/–: no portador). M: imagen superior, ecocardiograma con hipertrofia septal de 12mm (asterisco) y derrame pericárdico ligero (flecha); imagen inferior, engrosamiento de la válvula aórtica (flecha). 99mTc-DPD: tecnecio-99m con ácido-3,3-difosfono-1,2-propanodicarboxílico; ECG: electrocardiograma; RM: resonancia magnética; RTG: realce tardío de gadolinio; SGL: strain global longitudinal.")
Caso 1. A: electrocardiograma (ECG). B: cardiorresonancia magnética con realce tardío de gadolinio (RTG) mesocárdico septal anterior (flecha blanca). C: gammagrafía cardiaca con 99mTc-DPD con captación de grado 3. D: strain global longitudinal disminuido con conservación apical. Caso 2. E: ECG. F: ecocardiograma en plano apical de 4 cámaras que muestra engrosamiento concéntrico de ambos ventrículos. G: cardiorresonancia magnética con cinética alterada de gadolinio, RTG difuso y heterogéneo en los ventrículos izquierdo y derecho. H: gammagrafía cardiaca con 99mTc-DPD con captación de grado 3. Caso 3. I: ECG con fibrilación auricular, bajos voltajes y patrón de seudoinfarto. J: cardiorresonancia magnética con RTG difuso y cinética alterada de gadolinio. K: gammagrafía cardiaca con 99mTc-DPD con captación de grado 1-2. L: fragmento del árbol familiar (flecha: paciente 3; en negro, afectados de miocardiopatía por titina; cuadrado negro: varón afecto; círculo negro: mujer afecta; raya diagonal: paciente fallecido; cuadrado blanco: varón sano; N: normal; raya vertical: portador sano; E1 –/+: portador en heterocigosis TTN [p.Trp19433*]; E1 –/–: no portador). M: imagen superior, ecocardiograma con hipertrofia septal de 12mm (asterisco) y derrame pericárdico ligero (flecha); imagen inferior, engrosamiento de la válvula aórtica (flecha). 99mTc-DPD: tecnecio-99m con ácido-3,3-difosfono-1,2-propanodicarboxílico; ECG: electrocardiograma; RM: resonancia magnética; RTG: realce tardío de gadolinio; SGL: strain global longitudinal.
Estos 3 casos indican que el diagnóstico previo de una miocardiopatía en un paciente no debe hacer que se olvide la posibilidad de que pueda desarrollarse también una AC. Los 3 pacientes, con diagnósticos previos de miocardiopatía hipertrófica o miocardiopatía por titina, presentaron en su evolución RF clínicas y ecocardiográficas típicas de AC. Siempre deben tenerse en mente las RF de AC en pacientes mayores de 65-70 años, máxime en aquellos con HVI que presentan empeoramiento de su clínica habitual4–6, porque el diagnóstico de una miocardiopatía preexistente no debe suponer el fin de los procedimientos diagnósticos, ya que claramente ambas entidades pueden coexistir.
FINANCIACIÓNNo existe ninguna fuente de financiación asociada a esta investigación.
CONSIDERACIONES ÉTICASPor las características del trabajo, no ha sido necesaria la aprobación por el comité de ética local. Los autores confirman que se ha obtenido el consentimiento por escrito de los pacientes para la presentación y publicación de este artículo, incluidas las imágenes y sus textos correspondientes. Asimismo, se han tenido en cuenta las posibles variables de sexo y género de acuerdo con las directrices SAGER.
DECLARACIÓN SOBRE EL USO DE INTELIGENCIA ARTIFICIALNo se ha empleado ninguna herramienta de inteligencia artificial.
CONTRIBUCIÓN DE LOS AUTORESE. Martín-Álvarez, R. Barriales-Villa y J.M. Larrañaga-Moreira se han encargado del diseño, las figuras y la redacción del manuscrito. G. Barge-Caballero, M.G. Crespo-Leiro y B. Souto-Caínzos han colaborado en el análisis crítico.
CONFLICTO DE INTERESESJ. M. Larrañaga Moreira, M. G. Crespo Leiro y R. Barriales Villa declaran hacer recibido financiación de Pfizer para la asistencia a congresos.
M. G. Crespo Leiro ha recibido pagos de Pfizer a su institución por la participación en un ensayo clínico.
R. Barriales Villa ha participado como consultor de Pfizer, Alnylam y Akcea.