
Presentamos el caso de una familia con antecedentes de muerte súbita a edad temprana, inicialmente diagnosticada de taquicardia ventricular polimórfica catecolaminérgica (TVPC) con mutación p.L3778F en el gen del receptor de la rianodina (RyR2). Años después, mediante un completo estudio familiar y genético con Next Generation Sequencing (NGS), se identificó una segunda mutación patogénica en el gen KCNQ1 relacionada con síndrome de QT largo tipo 1 (SQTL1). Se demostró que la gravedad del fenotipo probablemente se debía a ambas mutaciones y no únicamente a p.L3778F en RyR2 como se había publicado previamente1.
El caso índice (III:5) sufrió una parada cardiorrespiratoria a los 8 años mientras nadaba, con recuperación completa tras reanimación. Su hermano de 10 años había muerto súbitamente también nadando (autopsia normal).
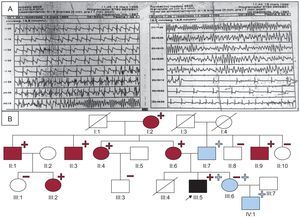
El electrocardiograma (ECG) del caso índice mostró bradicardia sinusal con QTc de 440 ms, mientras que el ecocardiograma, el Holter y la ergometría fueron normales. Dado el contexto de la parada cardiorrespiratoria se sospechó SQTL y se pautó tratamiento con bloqueadores beta (BB). Se realizó estudio electrofisiológico, sin inducción de arritmias y se implantó un Holter subcutáneo. A los 10 años, tras un esfuerzo físico, sufrió un síncope, y se observó una taquicardia ventricular polimórfica autolimitada (figura A).
 y/o E449R*14 en KCNQ1 (rojo);+: portadores en heterocigosis de L3778F en RyR2 (azul) y/o E449R*14 en KCNQ1 (rojo); círculo: mujer; cuadrado: varón; flecha: caso índice; relleno azul: paciente con TVPC (según consenso europeo y americano)3; relleno negro: clínicamente afectado de SQTL y TVPC; relleno rojo: paciente con SQTL; RyR2: receptor de la rianodina; SQTL1: síndrome de QT largo tipo 1; TVPC: taquicardia ventricular polimórfica catecolaminérgica. Esta figura se muestra a todo color solo en la versión electrónica del artículo.")
A: registro de la taquicardia ventricular polimórfica. B: árbol familiar./: fallecido; –: no portadores en heterocigosis de L3778F en RyR2 (azul) y/o E449R*14 en KCNQ1 (rojo);+: portadores en heterocigosis de L3778F en RyR2 (azul) y/o E449R*14 en KCNQ1 (rojo); círculo: mujer; cuadrado: varón; flecha: caso índice; relleno azul: paciente con TVPC (según consenso europeo y americano)3; relleno negro: clínicamente afectado de SQTL y TVPC; relleno rojo: paciente con SQTL; RyR2: receptor de la rianodina; SQTL1: síndrome de QT largo tipo 1; TVPC: taquicardia ventricular polimórfica catecolaminérgica. Esta figura se muestra a todo color solo en la versión electrónica del artículo.
Se derivó al paciente a un centro especializado en canalopatías para completar estudios y, tras realizar estudio genético mediante cribado mutacional (cromatografía líquida desnaturalizante de alto rendimiento [DHPLC]) y Sanger de los genes RyR2, KCNQ1, KCNH2, SCN5A, KCNE1 y KCNE2, se identificó una mutación missense en heterocigosis en RyR2 (p.L3778F). Se trató con BB, denervación simpática izquierda e implante de desfibrilador automático implantable. Se buscó la mutación en su madre, su padre y su hermana, todos asintomáticos, y se detectó en los dos últimos (figura B). La mutación no estaba descrita y el caso se publicó dentro de una serie de pacientes con TVPC, en la que se destacaba el mal pronóstico de los portadores varones1.
Años más tarde, la familia se trasladó de comunidad y el sobrino de 5 años del caso índice (IV:1) fue remitido a consulta para estudio. Estaba asintomático, pero se confirmó que era portador de la mutación familiar. Llamaba la atención que tanto su abuelo de 63 años (II:7) como su madre de 39 (III:6) fueran portadores y se mantuvieran asintomáticos con ECG, ergometría y Holter normales (excepto extrasistolia ventricular aislada de dos morfologías al inicio del ejercicio, que desapareció con el esfuerzo en una ergometría del abuelo).
Ante la sospecha de una segunda mutación, se realizó al caso índice un estudio genético mediante NGS (195 genes). Además de la mutación p.L3778F en RyR2, se detectó otra mutación en KCNQ1 (g.2610034_2610035insC, que ocasiona un transcrito aberrante con codón de parada en aminoácido 463, p.E449R*14), una mutación previamente publicada y relacionada con el SQTL12. Esta mutación no se había detectado en el estudio previo del paciente realizado mediante DHPLC/Sanger1.
Se continuó el estudio en colaboración con otros centros. La mutación en KCNQ1 se confirmó en 7 familiares (figura B), todos asintomáticos y con ECG basales con QTc medio de 460±15 ms (tabla). Según los actuales criterios diagnósticos, y dado que hasta un 25% de los pacientes con SQTL pueden tener en condiciones basales un QTc normal, los portadores de esta mutación patogénica se diagnosticaron de SQTL3.
Características de los portadores de las mutaciones en KCNQ1 y/o RyR2
| Caso | Sexo | Edad de diagnóstico (años) | Síncope | QTc (ms) | Mutación Glu449Arg*14 (KCNQ1) | Mutación Leu3778Phe (RyR2) |
|---|---|---|---|---|---|---|
| I:2 | M | 86 | No | 476 | + | – |
| II:1 | V | 67 | No | 446 | + | – |
| II:3 | V | 65 | No | 461 | + | – |
| II:4 | M | 64 | No | 474 | + | – |
| II:6 | M | 62 | No | 467 | + | – |
| II:9 | V | 57 | No | 483 | + | – |
| III:2 | M | 28 | No | 446 | + | – |
| II:7 | V | 63 | No | 432 | – | + |
| III:5 | V | 10 | Sí | 440 | + | + |
| III:6 | M | 39 | No | 416 | – | + |
| IV:1 | V | 5 | No | 406 | – | + |
M: mujer; RyR2: receptor de rianodina; V: varón.
Se pautaron BB a los portadores de ambas mutaciones, además de recomendar que se evitaran los fármacos que prolonguen el QT a los portadores de la mutación en KCNQ1. Hasta ahora todos permanecen asintomáticos, excepto el caso índice, único portador de ambas mutaciones, que sufrió a los 19 años una descarga apropiada del desfibrilador automático implantable.
En conclusión, en esta familia la mutación aislada p.L3778F en RyR2 probablemente no se relacione con un fenotipo tan grave como se ha publicado previamente aunque, hasta que no haya más estudios, no se puede descartar totalmente su patogenicidad, ya que la ergometría puede ser normal en pacientes afectados de TVPC4. La gravedad del caso índice y, probablemente en el hermano fallecido, se debería a la presencia de las dos mutaciones. El método DHPLC/Sanger puede dar falsos negativos y el estudio genético por NGS supera estas limitaciones5. Finalmente, se destaca la penetrancia incompleta y la expresividad variable del SQTL13,6, el valor del test genético para detectar a los portadores asintomáticos3,6 y la importancia de la colaboración entre centros para conseguir un estudio familiar completo.
FINANCIACIÓNEste artículo ha sido financiado parcialmente por fondos de la Red de Investigación Cardiovascular (RIC) (RD12/0042/0069, RD12/0042/0049).