
Las distroglicanopatías son un grupo heterogéneo de enfermedades de herencia autosómica recesiva (AR) con un amplio espectro clínico, que incluye la distrofia muscular congénita, la distrofia muscular de cinturas (DMC) y la miocardiopatía dilatada (MCD)1. Se caracterizan por una glicosilación reducida del α-distroglicano, esencial para la integridad muscular. La fukutina es una de las proteínas implicadas en su glucosilación y la fisiopatología de las distroglicanopatías2. Se presenta el primer caso descrito en Europa de un paciente con MCD y DMC por la variante genética p.Gly424Ser en homocigosis en el gen de la fukutina (FKTN), con demostración histopatológica de la alteración producida.
Un varón de 28 años consultó por debilidad y mialgias en las piernas, sin síntomas cardiacos. Presentaba pérdida de fuerza 4/5 en las extremidades inferiores, seudohipertrofia de tríceps y gemelos, atrofia de cuádriceps, signo de Gowers positivo (uso de las extremidades superiores para levantarse) y creatincinasa (CK) elevada (6.220 U/l). El electromiograma mostró afectación miopática moderada en las 4 extremidades. El electrocardiograma presentaba ritmo sinusal, PR corto y ondas Q y T negativas laterales y el ecocardiograma, una MCD con fracción de eyección del ventrículo izquierdo del 30%. Tras descartar enfermedad coronaria, se inició tratamiento con carvedilol y enalapril. El estudio se completó realizando un análisis genético del gen de la distrofina, que fue normal. Se obtuvo el consentimiento por escrito del paciente para la presentación y publicación de este artículo, incluidas las imágenes. También se recibió la autorización del comité de ética.
El paciente se mantuvo estable cardiológica y neurológicamente durante 15 años. A los 43 años, comenzó con disnea de moderados esfuerzos progresiva, e ingresó por insuficiencia cardiaca en situación de shock cardiogénico. El electrocardiograma (figura 1A) presentaba crecimiento biauricular, hemibloqueo anterior izquierdo y ondas Q laterales. El ecocardiograma (figura 1B) mostró MCD con disfunción grave (fracción de eyección del ventrículo izquierdo del 20%) y acinesia en segmentos inferolaterales. Recibió tratamiento neurohormonal optimizado e implante de desfibrilador automático. Tras una tórpida evolución, precisó trasplante cardiaco 6 meses después. En el estudio histológico, el corazón explantado presentaba extensa fibrosis biventricular.
. D: árbol familiar. FKTN: gen de la fukutina.")
Cinco años tras el trasplante, se mantenía estable de su miopatía y la CK continuaba elevada (figura 1C). Se le remitió a la unidad de cardiopatías familiares, donde se le realizó un estudio familiar. Los familiares de primer grado estaban sanos y sin miopatía, excepto su padre, a quien se le detectó una miocardiopatía hipertrófica (MCH) no obstructiva (grosor máximo septal, 16 mm). En un estudio genético realizado mediante secuenciación de nueva generación con un panel de 18 genes (figura 1D), no se encontró ninguna variante patogénica relacionada con MCH.
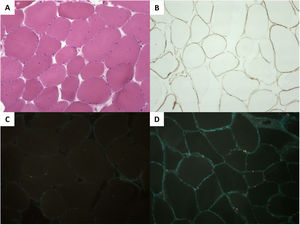
Se hizo un estudio genético con secuenciación de nueva generación al probando (panel de MCD, 96 genes) y se identificó la variante genética c.1270G>A, p.Gly424Ser en FKTN en homocigosis. Sus padres eran portadores heterocigotos. Una biopsia del músculo tríceps mostró leve afectación muscular, y en inmunohistoquímica se confirmó un déficit grave de α-distroglicano y leve de laminina α2; el resto del estudio fue normal (figura 2).
 fibras musculares con discreta variabilidad de diámetro y ocasionales núcleos internalizados. B: (inmunohistoquímica) leve reducción de la laminina α2. C: grave reducción del α-distroglicano en comparación con un control sano (D).")
Histopatología del músculo tríceps. A: (hematoxilina-eosina) fibras musculares con discreta variabilidad de diámetro y ocasionales núcleos internalizados. B: (inmunohistoquímica) leve reducción de la laminina α2. C: grave reducción del α-distroglicano en comparación con un control sano (D).
La fukutina es una ribitol 5-fosfato (Rbo5P) transferasa localizada en el aparato de Golgi, que trabaja con la proteína relacionada con la fukutina para incorporar parejas de Rbo5P al α-distroglicano, que se expresa principalmente en el músculo esquelético, el corazón y el cerebro2. Los portadores homocigotos o heterocigotos compuestos de variantes en FKTN presentan un amplio espectro fenotípico, consecuencia de la pérdida de función de la proteína. La FKTN se describió inicialmente en Japón a raíz de la distrofia muscular congénita de Fukuyama; se identificó como causa una variante fundadora en ese país y también se relacionó con el síndrome de Walker-Warburg: ambos cuadros producen un fenotipo grave con afectación cerebral y corta esperanza de vida. Posteriormente, se describieron casos en el resto del mundo, así como fenotipos más leves sin afectación cerebral como DMC (tipo R13). Generalmente, las variantes missense producen un fenotipo más leve, mientras que las no missense producen uno más grave, aunque hay excepciones3. La MCD forma parte del cuadro clínico a partir de la segunda década, y se ha descrito una forma infrecuente de MCD con afectación muscular escasa o ausente1,4,5. Parece que hay una disociación entre la afectación muscular y cardiaca; se ha descrito que la fukutina presenta otras funciones y que en el corazón es esencial para el mantenimiento de la contractilidad, la homeostasis cálcica, la integridad del aparato de Golgi y la resistencia de los miocardiocitos al estrés6.
La variante p.Gly424Ser se localiza en una región relevante para el proceso de fosforilación en el extremo C-terminal de FKTN. Aparece con baja frecuencia en la población de control (< 0,01%) en gnomAD, sin portadores homocigotos; el residuo afectado se encuentra muy conservado y la sustitución de glicina por serina se clasifica como patogénica por los predictores bioinformáticos. Se ha descrito recientemente, también en homocigosis, en 2 hermanos mexicanos con MCD, que fallecieron a los 20 y 21 años, sin aparente afectación neuromuscular, pero no se aporta información sobre la CK ni estudio histopatológico4.
Nuestro caso, primera descripción en Europa, confirma la probable patogenicidad de la variante p.Gly424Ser en FKTN, relacionada con una DMC leve en la que predomina la afectación cardiaca en forma de MCD. Este caso recalca la importancia de realizar un estudio genético de los pacientes con MCD y afección muscular e incluso en casos aislados, dada la posibilidad de que haya herencia AR.
FINANCIACIÓNNo se ha recibido financiación para la realización de este estudio.
CONTRIBUCIÓN DE LOS AUTORESJ.M. Larrañaga-Moreira y R. Barriales-Villa se han encargado del diseño, las figuras y la redacción del manuscrito. P. Blanco-Arias ha colaborado en la realización del estudio genético y el análisis crítico. B. San Millán-Tejado ha colaborado en la realización del estudio histológico, la figura 2, la redacción y el análisis crítico. G. Barge-Caballero y M.G. Crespo-Leiro han colaborado en la redacción y el análisis crítico.
CONFLICTO DE INTERESESP. Blanco-Arias es empleada de Health in Code S.L. Los demás autores no declaran ningún conflicto.