
El déficit primario de carnitina (DPC) es una enfermedad autosómica recesiva con una incidencia de 1/40.000-120.000 nacimientos. Lo causan mutaciones en el gen del transportador de cationes orgánico tipo 2 (OCTN2) de la membrana plasmática (SLC22A5), principalmente expresado en el miocardio. En consecuencia, las concentraciones plasmáticas de carnitina están reducidas (< 5μM; normal, 10-55μM) y la excreción urinaria está aumentada, lo que ocasiona una reducción de la transferencia de los ácidos grasos de cadena larga del citoplasma al interior de la mitocondria1.
La mayoría de los casos de DPC comunicados son lactantes y niños pequeños, frecuentemente con una afección multisistémica que incluye hepatomegalia, hipoglucemia hipocetósica, hiperamonemia, acidosis orgánica y encefalopatía hepática. Los escolares (5-6 años) suelen presentar debilidad muscular, aumento de transaminasas e insuficiencia cardíaca (miocardiopatía hipertrófica o dilatada)1,2.
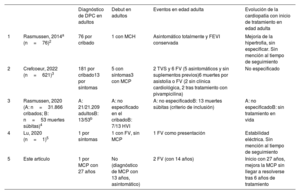
Pocos casos debutan en adolescentes y adultos jóvenes. En estas edades es posible que el diagnóstico y su tratamiento específico se retrasen años por la baja alerta de cardiólogos de adultos ante esta entidad. Una revisión sistemática reciente identificó a 161 sujetos diagnosticados por síntomas; de los 621 pacientes incluidos, solo 13 eran adultos (tabla 1). En ellos, la presentación suele ser exclusivamente cardíaca, y es frecuente la tríada miocardiopatía hipertrófica o dilatada, la no compactación y QT corto. En ausencia de un diagnóstico y un tratamiento adecuados, existe riesgo de arritmias ventriculares y muerte súbita (23/621, el 3,7% de los casos sintomáticos, 8 en edad adulta) o insuficiencia cardíaca grave3,4,5.
Resumen de casos de DPC en adultos
| Diagnóstico de DPC en adultos | Debut en adultos | Eventos en edad adulta | Evolución de la cardiopatía con inicio de tratamiento en edad adulta | ||
|---|---|---|---|---|---|
| 1 | Rasmussen, 2014a (n=76)2 | 76 por cribado | 1 con MCH | Asintomático totalmente y FEVI conservada | Mejoría de la hipertrofia, sin especificar. Sin mención al tiempo de seguimiento |
| 2 | Crefcoeur, 2022 (n=621)3 | 181 por cribado13 por síntomas | 5 con síntomas3 con MCP | 2 TVS y 6 FV (5 asintomáticos y sin suplementos previos)6 muertes por asistolia o FV (2 sin clínica cardiológica, 2 tras tratamiento con pivampicilina) | No especificado |
| 3 | Rasmussen, 2020 (A: n=31.866 cribados; B: n=53 muertes súbitas)4 | A: 21/21.209 adultosB: 13/53b | A: no especificado en el cribadoB: 7/13 HVI | A: no especificadoB: 13 muertes súbitas (criterio de inclusión) | A: no especificadoB: sin tratamiento en vida |
| 4 | Lu, 2020 (n=1)5 | 1 por síntomas | 1 con FV, sin MCP | 1 FV como presentación | Estabilidad eléctrica. Sin mención al tiempo de seguimiento |
| 5 | Este artículo | 1 por MCP con 27 años | No (diagnóstico de MCP con 13 años, asintomático) | 2 FV (con 14 años) | Inicio con 27 años, mejora la MCP sin llegar a resolverse tras 6 años de tratamiento |
DPC: déficit primario de carnitina; FEVI: fracción de eyección del ventrículo izquierdo; FV: fibrilación ventricular; HVI: hipertrofia ventricular izquierda; MCP: miocardiopatía; TVS: taquicardia ventricular sostenida.
En cuanto al tratamiento, la suplementación precoz con L-carnitina oral de 100-300mg/kg es eficaz en niños pequeños y consigue revertir rápida y completamente el fenotipo. Es un tratamiento seguro por tiempo indefinido, con seguimientos publicados a más de 30 años. Aun con normalización cardiológica, interrumpir los suplementos puede precipitar un deterioro clínico, recuperable si se retoman2,3. Frente a la evidencia acumulada acerca del beneficio del tratamiento precoz, contrasta la ausencia de publicaciones sobre su resultado instaurado en adultos muchos años tras la constatación de la miocardiopatía.
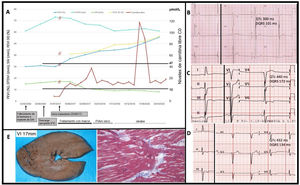
Se presenta el caso de DPC diagnosticado a los 27 años, tras años de tratamiento sintomático como miocardiopatía dilatada con hipertrofia, no compactación y QT acortado. El paciente inició controles tras la muerte súbita de su hermana con 2 intentos de estudio genético negativos. Al poco tiempo se le implantó un desfibrilador en prevención primaria que dio 2 terapias apropiadas por fibrilación ventricular a los 6 meses. Catorce años después, con el diagnóstico de DCP clínico (cifras de carnitina muy bajas) y genético (heterocigoto compuesto para las variantes patogénicas SLC22A5 p.Glu452Lys y p.Leu269HisfsTer27) inició tratamiento de sustitución ajustado a peso (marca comercial, seguido de fórmulas magistrales con polvo seco inicialmente y con jarabe después, con el que se consiguieron valores más estables y en rango normal). La mejoría del intervalo QT fue muy rápida, y no se han detectado eventos arrítmicos. Tanto la función sistólica como los diámetros ventriculares han mejorado, sin normalizarse completamente, a diferencia de lo comunicado con el tratamiento precoz y en la infancia (figura 1). La no compactación se ha mantenido inalterada. Los padres y un hermano sanos resultaron heterocigotos simples.
 e histológico (foco de disarray) de la autopsia de la hermana fallecida.")
A: paciente con DPC y tratamiento sustitutivo iniciado con 27 años, tras 14 años de retraso desde el debut de la cardiopatía. B–D: electrocardiograma al diagnóstico, al poco del debut y tras 5 años de tratamiento. E: imagen del estudio macroscópico (hipertrofia) e histológico (foco de disarray) de la autopsia de la hermana fallecida.
El diagnóstico y el tratamiento del DPC en adultos entrañan una serie de retos que se van a exponer.
A menudo el diagnóstico presintomático se alcanza en el cribado neonatal sistemático de enfermedades endocrinometabólicas, si bien el DPC no se incluía en estos programas cuando muchos de los adultos actuales nacieron, no se incluye de manera homogénea en diferentes regiones y tiene reconocidos pros y contras.
Esta entidad, muy desconocida entre cardiólogos, se puede tratar como la miocardiopatía de base, sin el tratamiento etiológico sustitutivo que podría revertir o soslayar el fenotipo y sin poder darle el adecuado enfoque familiar.
Algunos paneles genéticos comerciales no incluyen el gen SLC22A5 y otros lo añadieron entre 2015 y 2020, por lo que estudios más antiguos no lo analizaron.
Tras el diagnóstico, los adultos requieren altas dosis de carnitina, ya que se ajustan por el peso corporal. A esas dosis es frecuente el efecto secundario del mal olor corporal (a pescado podrido).
La monitorización debe ser regular para confirmar que la concentración de carnitina libre se encuentra en el intervalo normal aun en ausencia de mal cumplimiento o inestabilización clínica.
No se ha publicado evidencia sobre la efectividad y la seguridad del tratamiento sustitutivo iniciado tras muchos años de haberse constatado el fenotipo miocardiopático.
En respuesta a todos estos retos, se plantea que:
- •
Conviene recordar que pueden llegar casos sin cribado neonatal ni diagnóstico a la edad adulta y debutar con cardiopatía.
- •
Debe incrementarse la sensibilidad de los cardiólogos de adultos frente a esta entidad a fin de mejorar su sospecha y confirmación.
- •
La evolución de los estudios cardiogenéticos con número creciente de genes (estudios de exoma y genoma) permitirán analizar sistemáticamente también genes minoritarios, como SLC22A5. Mientras se utilicen paneles de genes, se aconseja estar atentos a la tríada clínica miocardiopatía hipertrófica o dilatada, no compactación y QT corto para añadir expresamente el gen SLC22A5.
- •
Las grandes dosis de L-carnitina requeridas en adolescentes y adultos indefinidamente motivan evitar la marca comercial (necesidad de muchas ampollas y alto precio) y seleccionar fórmulas magistrales más concentradas preparadas en farmacia hospitalaria con formulación en polvo seco o jarabe. En nuestra experiencia, el jarabe ha sido la solución para lograr valores normales estables.
- •
El olor corporal desagradable por el sobrecrecimiento bacteriano intestinal puede combatirse con metronidazol 10mg/kg/día, 10 días al mes6.
- •
Para facilitar el seguimiento, pueden intercalarse citas no presenciales, en las que los pacientes remiten por correo directamente al laboratorio el papel Whatman con las 3 gotas de sangre requeridas para determinar la carnitina libre.
Nuestra experiencia y una revisión de la literatura nos han permitido plasmar aquí unos consejos útiles para mejorar el diagnóstico y el tratamiento de estos pacientes. Por último, se destaca que la expectativa de curación completa no es tan alta como la actualmente descrita en niños de corta edad cuando el inicio de tratamiento en adultos se demora años respecto a la aparición del fenotipo cardiaco.
FINANCIACIÓNLa presente investigación no ha recibido ayudas específicas provenientes del sector público ni del privado.
CONSIDERACIONES ÉTICASEl trabajo ha sido aprobado por el Comité Ético del Hospital Universitario y Politécnico La Fe de Valencia.
Se obtuvo el consentimiento informado de los pacientes para la publicación de su caso.
El presente trabajo se adecua a las directrices SAGER.
DECLARACIÓN SOBRE EL USO DE INTELIGENCIA ARTIFICIALNo se ha empleado inteligencia artificial para el desarrollo del presente trabajo.
CONTRIBUCIÓN DE LOS AUTORESTodos los autores contribuyeron en mayor o menor medida a todas las tareas siguientes: contribuir sustancialmente a la concepción y el diseño del trabajo, adquisición de datos o su análisis e interpretación; redactar el artículo o hacer una revisión crítica de su contenido intelectual; dar la aprobación final a la versión que se publicará, y acceder a asumir responsabilidades sobre todos los aspectos del artículo y a investigar y resolver cualquier cuestión relacionada con la exactitud y veracidad de cualquier parte del trabajo.
CONFLICTO DE INTERESESNinguno.
AGRADECIMIENTOSAgradecemos la desinteresada participación de la familia, el servicio de Análisis Clínicos-Metabolopatías de nuestro centro y el Instituto de Medicina Legal de Valencia. Las muestras incluidas fueron gestionadas por Biobanco La Fe (B.0000723) y secuenciadas en la Unidad de Genómica del Instituto de Investigación Sanitaria La Fe de Valencia.