Palabras clave
INTRODUCCIÓN
El origen anómalo de una rama de la arteria pulmonar desde la aorta es una cardiopatía congénita infrecuente, que consiste en el nacimiento de una de las ramas pulmonares de la aorta ascendente. Incluida en las anomalías del arco aórtico, está producida por un fallo parcial o completo del desarrollo del sexto arco izquierdo1.
Debe sospecharse en recién nacidos con fallo cardíaco y aumento del flujo pulmonar. Sin cirugía es mortal en la mayoría de las ocasiones, debido a la hipertensión pulmonar irreversible, que puede estar presente a edades muy tempranas2,3.
Describimos el caso de un recién nacido diagnosticado prenatalmente de tetralogía de Fallot, que presentó en su evolución fallo cardíaco. El cateterismo demostró tetralogía de Fallot asociada al origen anómalo de una rama de la arteria pulmonar (AP) desde la aorta izquierda desde la aorta ascendente. Se realizó una intervención quirúrgica definitiva con buen curso posterior.
CASO CLÍNICO
Neonato a término con diagnóstico prenatal de tetralogía de Fallot. La ecocardiografía posnatal demostró una comunicación interventricular subaórtica amplia con cabalgamiento aórtico del 50%, tronco de la AP de 8 mm y ramas pulmonares de 4 mm; no pudo verse el origen anómalo de una rama de la arteria pulmonar desde la aorta (fig. 1B y 1D), no había estenosis valvular ni subvalvular pulmonar. La segunda semana de vida presentó en forma progresiva taquipnea, taquicardia, hepatomegalia y soplo sistólico II/VI en mesocardio. La radiografía de tórax demostró cardiomegalia, edema pulmonar y punta del corazón elevada. Se inició tratamiento con digitálicos y diuréticos, con mejoría parcial.

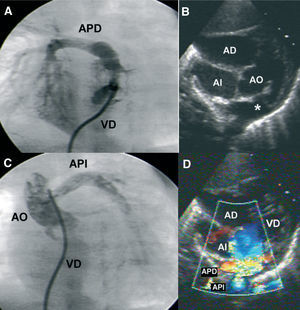
Fig. 1. A: ventriculografía derecha; se observa tronco de la arteria pulmonar (AP), AP derecha y ausencia de la AP izquierda. B: corte subcostal; se observa la comunicación interventricular, estenosis infundibular pulmonar. Véase la falsa imagen de ambas ramas emergiendo del tronco de la AP (*). C: aortografía ascendente; se observa la opacificación de la AP izquierda que nace de la cara lateral de la aorta. D: el mismo corte pero con Doppler color. AD: aurícula derecha; AI: aurícula izquierda; AO: aorta; APD: arteria pulmonar derecha; API: arteria pulmonar izquierda; VD: ventrículo derecho.
Dado que la clínica no era compatible con los hallazgos ecocardiográficos, se realizó cateterismo cardíaco. El ventrículo derecho (VD) tenía buena función, había un gradiente infundíbulo-valvular de 20 mmHg, tronco de AP con ausencia de AP izquierda (fig. 1A), hipertensión pulmonar por hiperaflujo (Qp/Qs 2,4) con resistencias normales; ambos ventrículos tenían las mismas presiones. En la aortografía se apreció arco aórtico derecho, nacimiento de la AP izquierda de la cara posterolateral de la aorta ascendente (fig. 1C). La ventriculografía izquierda mostró una comunicación interventricular subaórtica amplia.
El estudio para microdeleción 22q11 fue positivo. A los 23 días de vida se realizó una corrección quirúrgica; se practicó la extracción de la AP izquierda y el reimplante sobre el tronco de la AP, el cierre directo de la aorta ascendente y de la comunicación interventricular subaórtica con parche de pericardio heterólogo. Se hizo resección del músculo infundibular y de bandas anómalas del VD por vía auricular. Se realizó una ecocardiografía postoperatoria que mostró gradiente de 17 mmHg a la altura de la AP izquierda y presiones en VD al 40% de las sistémicas. Una semana más tarde fue dado de alta.
DISCUSIÓN
El origen anómalo de una rama de la AP desde la aorta es una malformación conotruncal muy rara, que podría suponer el 0,12% de las cardiopatías congénitas4. La rama anómala se origina la mayoría de las ocasiones en la cara posterolateral de la aorta ascendente, cerca de la válvula aórtica5,6. En el 15% se origina de forma distal, cerca de la base de la arteria innominada4. El origen anómalo de la rama derecha es 5-6 veces más frecuente que el de la izquierda4,6,7.
La mayoría de los artículos sobre el origen anómalo de una rama de la AP desde la aorta presentan casos clínicos aisladas salvo 26,7 donde se revisan 12 y 16 casos, respectivamente.
El origen anómalo de una rama de la AP desde la aorta puede presentarse de forma aislada o asociado a otros defectos cardíacos congénitos; el más frecuente es la persistencia conducto arterioso3,4,6. Cuando la rama anómala es la izquierda, la cardiopatía asociada más frecuente es la tetralogía de Fallot8. La asociación a microdeleción del cromosoma 22q11, como sucedió en nuestro paciente, sólo se ha descrito en un caso, lo que demuestra que esta anomalía pertenece al conjunto de malformaciones cardíacas conotruncales debidas a un desarrollo anómalo de las células de la cresta neural9.
Se han descrito sólo 9 casos de asociación de origen anómalo de una rama de la AP desde la aorta izquierda y tetralogía de Fallot; el caso presentado es el primero en la literatura médica española.
La presentación clínica incluye fallo cardíaco, soplos cardíacos, taquipnea, disnea e infecciones respiratorias de repetición. La insuficiencia cardíaca es el cuadro predominante, y es especialmente grave cuando se asocia a coartación o interrupción del arco aórtico10.
La ecocardiografía bidimensional con Doppler color es el método diagnóstico inicial, aunque puede pasar desapercibida hasta en un 15% de casos10. En nuestro caso no pudimos detectar el nacimiento anómalo probablemente porque ambas ramas estaban muy próximas, tal como se observa en la figura 1 (B y D). La exploración con Doppler puede ser sugestiva de nacimiento anómalo11, objetivándose un flujo sistodiastólico en la AP izquierda, fenómeno que no observamos probablemente por la elevada resistencia vascular pulmonar propia del recién nacido. El cateterismo cardíaco sigue siendo la prueba para el diagnóstico final, que además es útil para planificar la cirugía.
El cateterismo demuestra presiones pulmonares sistémicas en ambas arterias. El pulmón anormalmente conectado es perfundido a una presión sistémica, mientras que el otro está expuesto a todo el gasto cardíaco del VD. Cuando en el período neonatal caen las resistencias pulmonares, la presión y el flujo dentro de la AP anómala se incrementan, lo que causa una sobrecirculación y lleva a una hipertensión pulmonar10. El desarrollo de hipertensión pulmonar en el pulmón que se irriga normalmente del tronco de la arteria pulmonar podría deberse a sustancias circulantes vasoconstrictoras o factores neurogénicos cruzados12,13.
La cirugía temprana, en edad neonatal, es la preferida11 para evitar la enfermedad vascular oclusiva pulmonar que puede desarrollarse de forma acelerada a partir de los 3 meses2,3. La cirugía consiste en la anastomosis directa de la rama anómala al tronco de la arteria pulmonar3,11. Puede ser necesario ampliar la zona intervenida con parche de pericardio autólogo o la interposición de un homoinjerto11. Quizá las técnicas que empleen tejidos autólogos para ensanchar y alargar el origen anómalo de una rama de la AP desde la aorta estén asociadas a una menor reestenosis que las técnicas de anastomosis directa14, aunque no hay series con suficientes pacientes que lo puedan corroborar. La mortalidad es en la actualidad prácticamente inexistente.
La complicación quirúrgica postoperatoria más frecuente es la estenosis de la anastomosis (10,6%), que puede desarrollarse meses después de la cirugía7. Los pacientes con reestenosis severa pueden requerir angioplastia quirúrgica, dilataciones con balón o implantación de stents7.
El origen anómalo de una rama de la AP desde la aorta es una rara entidad en la que un rápido diagnóstico y una pronta cirugía son esenciales para prevenir la enfermedad pulmonar vascular irreversible. Es importante el seguimiento para detectar la frecuente reestenosis.
Correspondencia: Dr. F. Prada.
Sección de Cardiología Pediátrica. Hospital Sant Joan de Déu.
Passeig de Sant Joan de Déu, 2. 08950 Esplugues. Barcelona. España.
Correo electrónico: fprada@hsjdbcn.org