
Las mutaciones en el gen de la troponina T (TNNT2) se han asociado en pequeños estudios al desarrollo de miocardiopatía hipertrófica caracterizada por alto riesgo de muerte súbita e hipertrofia leve. Se describe el curso clínico de los pacientes portadores de mutaciones en este gen.
MétodosSe analizaron las características clínicas y el pronóstico de los sujetos con mutaciones en el gen TNNT2 atendidos en una unidad de cardiopatías familiares.
ResultadosA partir de 180 familias con miocardiopatías estudiadas genéticamente, se identificó a 21 (11,7%) con mutaciones en TNNT2: 10 familias Arg92Gln, 5 Arg286His, 3 Arg278Cys, 1 Arg92Trp, 1 Arg94His y 1 Ile221Thr. A través de la evaluación familiar se identificó a 33 portadores genéticos adicionales. El estudio incluyó a 54 portadores genéticos: el 56% varones con una media de edad de 41±17 años; 33 miocardiopatías hipertróficas, 9 dilatadas y 1 no compactada, con grosor máximo de 18,5±6mm; con disfunción ventricular el 30% y antecedentes de muerte súbita el 62%. En el seguimiento 4 fallecieron y 14 (33%) recibieron un desfibrilador (8 probandos, 6 familiares). La supervivencia media fue de 54 años. Los portadores de Arg92Gln tuvieron desarrollo precoz, alta penetrancia, alto riesgo de muerte súbita, alta tasa de implante de desfibrilador y alta frecuencia de fenotipo mixto.
ConclusionesLas mutaciones en el gen TNNT2 fueron más frecuentes en esta serie. Su perfil clínico y pronóstico depende de la mutación hallada. Los portadores de la mutación Arg92Gln desarrollaron miocardiopatía hipertrófica o dilatada y tuvieron un pronóstico significativamente peor que con otras mutaciones en TNNT2 u otros genes sarcoméricos.
Palabras clave
La miocardiopatía hipertrófica (MCH) es una enfermedad hereditaria autosómica dominante con presentación clínica e historia natural muy heterogéneas1, y causa frecuente de muerte súbita cardiaca (MSC) de jóvenes2–4; se asocia a mutaciones en genes que codifican proteínas del sarcómero5–7. En la literatura se debate sobre la correlación genotipo-fenotipo de mutaciones individuales7,8, y se trata de establecer un pronóstico, según la mutación hallada, que pueda ayudar a estratificar la enfermedad y dar un adecuado consejo genético a las familias. Las mutaciones en el gen de la troponina T (TNNT2) se describieron hace años en algunas publicaciones con escaso número de familias, y ya se postuló una alta prevalencia de MSC en jóvenes portadores5,6,9,10 que, además, presentaban un fenotipo de hipertrofia ventricular izquierda leve6,11.
El objetivo del estudio es describir el curso clínico en una serie de pacientes y familiares —relativamente amplia para la baja prevalencia de la enfermedad— portadores de mutaciones en TNNT2 y ampliar los datos conocidos hasta ahora sobre su pronóstico.
MÉTODOSLa cohorte está formada por probandos aparentemente no relacionados y afectados de miocardiopatía, la mayoría con fenotipo de MCH, evaluados en una consulta de cardiopatías familiares en el Hospital Son Llàtzer (Palma de Mallorca, Islas Baleares, España) durante un periodo de 7 años, a los que se realizó estudio genético de mutaciones en el gen TNNT2 (y también otros cuatro genes sarcoméricos: MYBPC3, MYH7, TNNI3 y TPM1, e incluso lamina A/C si el fenotipo del probando era miocardiopatía dilatada [MCD]). A todos los familiares de los portadores se les propuso realizar una evaluación clínica y genética.
La MCH se diagnostica cuando el grosor máximo miocárdico (GMM) es ≥ 15mm en al menos un segmento en ausencia de otras enfermedades que expliquen la hipertrofia12,13. En probandos con MSC como primera manifestación clínica, el diagnóstico de MCH se confirmó en necropsia siempre que fue posible. Se consideró afectados a los familiares cuando cumplían los criterios familiares de MCH (≥ 13mm)14.
A todos los pacientes y familiares se les realizó electrocardiograma, ecocardiograma, ergometría y Holter electrocardiográfico de 24h, de acuerdo con los métodos descritos15, y una cardiorresonancia siempre que fue posible.
Los principales factores de riesgo de MSC se definieron como: antecedentes familiares de MSC, episodio de síncope de origen arrítmico o etiología desconocida, taquicardia ventricular no sostenida ≥ 120 lpm, GMM ≥ 30mm y respuesta anormal de la presión arterial en ejercicio (menores de 40 años)16.
El gen TNNT2 se secuenció con Sanger o next-generation sequencing (NGS). De los 21 probandos, se estudió a 19 mediante Sanger (se secuenciaron los cinco genes sarcoméricos principales: MYBPC3, MYH7, TNNT2, TNNI3, TPM1) y a 2 por NGS (en un caso 12 genes: cinco sarcoméricos más ACTC1, GLA, MYL2, MYL3, PRKAG2, PTPN11 y TNNC1, y en otro 27 genes: los 12 previos más CASQ2, DMD, DTNA, FKBP1A, KCNH2, LDB3, LMNA, MIB1, MYH6, NOTCH1, PLN, RYR2, SCN5A, TAZ y TTN, este último por fenotipo de miocardiopatía no compactada). Un cambio en la secuencia de aminoácidos en comparación con la secuencia de referencia se consideró mutación patógena cuando presentaba los siguientes criterios: segregaba con los miembros afectados de la familia, no estaba presente en 200 cromosomas de individuos sanos no relacionados, no se había identificado hasta la fecha en poblaciones de miles de individuos de diferentes grupos étnicos incluidos en el Proyecto 5.000 Genomas (Exome Variant Server), 1.000 Genomas y dbSNP (Short Genetic Variations database), y afectaban a un residuo filogenéticamente conservado entre especies e isoformas de troponina T. Se consideró variante alélica rara cuando no se pudo demostrar la segregación y no estaba presente en los controles, y se consideró polimorfismo siempre que no se asociara con la enfermedad y estuviera presente en los controles. Las variantes descritas previamente se revisaron para evaluar su patogenicidad, y las nuevas mutaciones se estudiaron con herramientas in silico.
Se obtuvo consentimiento informado para extracción de ADN de cada individuo. El estudio respetó los principios de la Declaración de Helsinki, el Convenio del Consejo de Europa sobre Derechos Humanos y Biomedicina y la Declaración Universal sobre el Genoma Humano y los Derechos Humanos de la UNESCO.
El análisis estadístico se realizó utilizando la aplicación SPSS (v.15.0, SPSS Inc.; Chicago, Illinois, Estados Unidos). Los datos distribuidos normalmente se expresan como media (intervalo de confianza del 95%). Las diferencias entre las medias se compararon mediante la prueba de la t de Student de dos colas no apareadas. La prueba de la χ2 se utilizó para comparar datos categóricos. Se utilizó la prueba de la U de Mann-Whitney para analizar datos continuos sin distribución normal. Los resultados predefinidos en el análisis de supervivencia fueron: MSC, primer choque apropiado del desfibrilador automático implantable (DAI), muerte por insuficiencia cardiaca, trasplante cardiaco u otra muerte cardiovascular. La probabilidad acumulada para la ocurrencia de un evento se calculó utilizando el método de Kaplan-Meier. Se realizó una comparación con los datos publicados anteriormente. El análisis de supervivencia desde el nacimiento también se realizó para las mutaciones individuales. Se consideró estadísticamente significativo un valor de probabilidad de p<0,05.
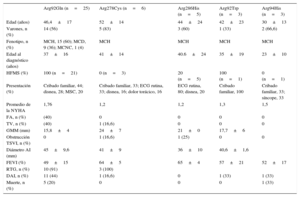
RESULTADOSSe estudió a 180 probandos, consecutivos y no relacionados, afectados de miocardiopatía (155 MCH, 15 MCD y 10 no compactadas) en busca de mutaciones en el gen TNNT2 y en los otros cuatro genes principales del sarcómero (MYBPC3, MHY7, TNNI3 y TPM1); 21 (11,7%) tenían mutaciones patógenas en TNNT2; 98 familiares dieron su consentimiento para el examen clínico (media, 4,7 familiares/familia) y 78 para el análisis genético de TNNT2; en 33 (42%) se halló alguna mutación. La suma de probandos y familiares portadores de mutaciones en TNNT2 fue de 54. Asumiendo que algunos familiares de primer grado sin estudio genético pero afectados de MCH tendrían la misma mutación en TNNT2, la suma total fue de 68 pacientes: 57 afectados y 11 portadores asintomáticos.
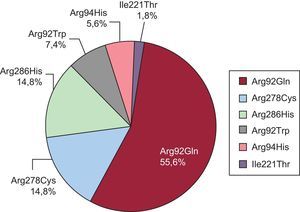
Se identificaron seis mutaciones diferentes en 21 familias: Arg92Gln en 10 familias, Arg286His en 5, Arg278Cys en 3 y Arg92Trp, Arg94His e Ile221Thr en una familia cada una. Todas estas variantes excepto una (Ile221Thr) ya se han publicado como causantes de MCH8,14,15,17–19.
Se encontraron dobles mutaciones en 4 probandos (19%): 2 con la mutación Arg278Cys (1 con la mutación Arg502Gln en MYBPC3 y 1 con Arg723Cys en MHY7), 1 probando con la mutación Arg286His y la mutación Arg326Gln en MYBPC3 y 1 probando con la mutación Arg92Gln, portador asimismo de una variante en MYBPC3 que podría actuar de modificador genético. Ningún familiar era portador de dobles mutaciones. La distribución de mutaciones y pacientes fue la siguiente (figura 1): 30 Arg92Gln (55,6%), 8 Arg278Cys (14,8%), 8 Arg286His (14,8%), 4 Arg92Trp (7,4%), 3 Arg94His (5,6%) y 1 Ile221Thr (1,8%).

A todos los pacientes afectados se les realizó una estratificación del riesgo de MSC. Tenían antecedentes de MSC 13 probandos (62%), el 100% de las familias (n=10) con la mutación Arg92Gln.
Se estudiaron el electrocardiograma y el ecocardiograma en la primera evaluación de todos los portadores de la mutación, excepto 3 pacientes (por MSC como presentación clínica). El electrocardiograma fue habitualmente anormal (criterios de voltaje de hipertrofia ventricular izquierda con ondas T negativas en precordiales e inferiores), a veces con anormalidades solo leves en el ecocardiograma.
De los pacientes con la mutación Arg92Gln, 9 tenían un fenotipo de MCD en la primera evaluación, con disfunción ventricular izquierda grave. Además, un individuo afectado con Arg92Gln tenía un fenotipo de miocardiopatía no compactada. El resto presentaba MCH (tablas 1 y 2).
Datos clínicos y genéticos de las familias y pacientes incluidos en el estudio
| Familia | Mutación TNNT2 | Edad/sexo | Fenotipo | Edad al diagnóstico (años) | HFMS | Presentación | Clase funcionalde la NYHA | FA | TV | GMM (mm) | Obstrucción TSVI | Diámetro AI (mm) | FEVI | RTG | MP/DAI (edad, años) | Terapia apropiada DAI | Muerte |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1/Probando | Arg92Gln | 22/V | MCD | 22 | Sí | MSC | I | No | No | 12 | No | No | Sí | ||||
| 1/Madre | Arg92Gln | 70/M | MCD | 60 | Disnea | II | Sí | No | 11 | No | 44 | 40 | MP (60) | No | |||
| 1/Hermana | Arg92Gln | 42/M | MCD | 42 | MSC | I | Sí | No | 8 | No | No | Sí | |||||
| 2/Probando | Arg92Gln | 31/V | MCH | 13 | Sí | MSC | I | No | Sí | 20 | No | 48 | 60 | DAI (13) | Sí | No | |
| 2/Padre | Arg92Gln | 53/V | MCH | 37 | Cribado familiar | II | Sí | Sí | 20 | No | 52 | 55 | DAI (38) | Sí | No | ||
| 2/Tía | Arg92Gln | 45/M | MCH | 28 | Cribado familiar | III | Sí | No | 22 | No | 50 | 60 | Sí | No | No | ||
| 3/Probando | Arg92Gln | 64/M | MCD | 50 | Sí | Disnea | II | Sí | No | 14 | No | 50 | 45 | Sí | No | No | |
| 3/Hermana | Arg92Gln | 40/M | MCH | 34 | Cribado familiar | I | No | No | 14 | No | 46 | 60 | Sí | No | No | ||
| 3/Nieto* | Arg92Gln | 8/V | — | — | I | No | No | 7 | No | 25 | 65 | No | No | ||||
| 3/Hermano | Arg92Gln | 76/V | MCD | 69 | Disnea | IV | Sí | Sí | No | 30 | No | Sí | |||||
| 3/Sobrino | Arg92Gln | 45/V | MCH | 35 | Cribado familiar | I | No | No | 17 | No | 33 | 48 | Sí | No | No | ||
| 3/Sobrina | Arg92Gln | 52/M | MCH | 43 | Cribado familiar | I | No | No | 14 | No | 45 | 60 | No | No | |||
| 3/Sobrino-nieto | Arg92Gln | 21/V | MCH | 20 | Cribado familiar | I | No | No | 18 | No | 65 | Sí | No | Sí | |||
| 3/Sobrina-nieta | Arg92Gln | 19/M | MCH | 19 | Cribado familiar | I | No | No | 14 | No | 62 | Sí | DAI (19) | No | No | ||
| 4/Probando | Arg92Gln | 72/V | MCD | 67 | Sí | Disnea | III | Sí | No | 11 | No | 55 | 30 | Sí | TRC (69) | Sí | |
| 4/Hermana | Arg92Gln | 65/M | MCD | 55 | Disnea | III | Sí | Sí | 15 | No | 60 | 33 | DAI (63) | ? | No | ||
| 4/Sobrino | Arg92Gln | 54/V | MCD | 50 | Disnea | III | No | No | No | 30 | TRC (52) | No | |||||
| 4/Sobrino-nieto | Arg92Gln | 19/V | MCH | 11 | MSC | I | No | Sí | 13 | No | 30 | 65 | DAI (11) | ? | No | ||
| 5/Probando | Arg92Gln | 66/V | MCD | 40 | Sí | Dolor torácico | II | Sí | Sí | 20 | No | 50 | 30 | DAI + TRC (62) | ? | No | |
| 6/Probando | Arg92Gln | 49/V | MCH | 35 | Sí | Cribado familiar | I | No | Sí | 15 | No | 43 | 60 | Sí | DAI (45) | No | No |
| 6/Hermana | Arg92Gln | 54/M | MCH | 24 | Cribado familiar | II | Sí | Sí | 19 | No | 48 | 30 | DAI | ? | No | ||
| 6/Hija* | Arg92Gln | 7/F | — | — | I | No | No | 7 | No | 27 | 70 | No | No | ||||
| 7/Probando | Arg92Gln | 40/V | MCH | 36 | Sí | Cribado familiar | I | No | No | 22 | No | 47 | 60 | Sí | DAI (38) | No | No |
| 7/Sobrino* | Arg92Gln | 12/V | — | — | I | No | No | 8 | No | 28 | 65 | No | No | ||||
| 7/Sobrina* | Arg92Gln | 6/M | — | — | I | No | No | No | 60 | No | No | ||||||
| 8/Probando | Arg92Gln | 36/V | MCH | 17 | Sí | MSC | I | No | Sí | 20 | No | 33 | 62 | DAI (17) | ? | No | |
| 8/Hijo* | Arg92Gln | 8/V | — | — | I | No | No | 9 | No | 27 | 70 | No | No | ||||
| 9/Probando | Arg92Gln | 52/M | MCH | 51 | Sí | Disnea | II | No | No | 15 | No | 31 | 58 | No | No | No | |
| 9/Hija | Arg92Gln | 27/M | MCH | 26 | Cribado familiar | I | No | No | 20 | No | 29 | 65 | Sí | No | No | ||
| 10/Probando | Arg92Gln | 46/V | MCNC | 40 | Sí | ECG de rutina | III | No | Sí | 10 | No | 60 | 21 | DAI (44) | No | No | |
| 11/Probando | Arg92Trp | 69/M | MCH | 57 | Sí | Cribado familiar | II | No | No | 25 | No | 42 | 33 | DAI (69) | No | No | |
| 11/Hijo | Arg92Trp | 32/V | MCH | 28 | Cribado familiar | I | No | No | 15 | No | 40 | 65 | No | No | |||
| 11/Hija | Arg92Trp | 25/M | MCH | 20 | Cribado familiar | I | No | No | 13 | No | 40 | 74 | Sí | No | No | ||
| 11/Hija* | Arg92Trp | 36/M | — | — | I | No | No | 10 | No | 33 | 65 | No | No | ||||
| 12/Probando | Arg278Cys | 47/V | MCH | 18 | No | ECG de rutina | I | No | No | 23 | No | 38 | 60 | Sí | No | No | |
| 12/Padre | Arg278Cys | 71/V | MCH | 57 | ECG de rutina | I | No | No | 35 | Sí | 58 | 58 | No | No | |||
| 12/Primo | Arg278Cys | 40/V | MCH | 37 | Cribado familiar | I | No | No | 16 | No | 36 | 62 | No | No | |||
| 13/Probando | Arg278Cys | 41/M | MCH | 37 | No | Disnea | I | No | No | 18 | No | 35 | 65 | Sí | No | No | |
| 13/Hermano | Arg278Cys | 44/V | MCH | 42 | Cribado familiar | I | No | No | 22 | No | 36 | 70 | Sí | No | No | ||
| 13/Madre* | Arg278Cys | 75/M | — | — | I | No | No | 10 | No | 32 | 60 | No | No | ||||
| 14/Probando | Arg278Cys | 68/V | MCH | 55 | No | Dolor torácico | II | No | Sí | 30 | No | 44 | 68 | DAI (62) | Sí | No | |
| 14/Hija* | Arg278Cys | 29/M | — | — | I | No | No | 9 | No | 31 | 62 | No | No | ||||
| 15/Probando | Arg286His | 80/M | MCH | 77 | Sí | Disnea | II | No | No | 21 | Sí | 51 | 70 | MP (79) | No | ||
| 15/Hijo* | Arg286His | 48/V | — | — | I | No | No | 10 | No | 35 | 65 | No | No | ||||
| 16/Probando | Arg286His | 35/V | MCH | 25 | No | ECG de rutina | I | No | No | 21 | No | 34 | 60 | No | No | ||
| 17/Probando | Arg286His | 36/M | MCH | 34 | No | ECG de rutina | I | No | No | No | No | ||||||
| 18/Probando | Arg286His | 16/V | MCH | 15 | No | ECG de rutina | I | No | No | 21 | No | 28 | 65 | No | No | ||
| 18/Madre* | Arg286His | 40/M | — | — | I | No | No | 10 | No | 35 | 60 | No | No | ||||
| 18/Primo* | Arg286His | 6/V | — | — | I | No | No | No | 75 | No | No | ||||||
| 19/Probando | Arg286His | 53/V | MCH | 52 | No | ECG de rutina | I | No | No | 21 | No | 32 | 67 | Sí | No | No | |
| 20/Probando | Arg94His | 15/V | MCH | 14 | Sí | Cribado familiar | I | No | No | 33 | No | 41 | 64 | No | No | No | |
| 20/Padre | Arg94His | 39/V | MCH | 34 | Síncope | II | No | No | No | 40 | DAI (34) | ? | No | ||||
| 20/Tía | Arg94His | 36/M | MCH | 22 | No | Sí | |||||||||||
| 21/Probando | Ile221Thr | 75/M | MCH | 62 | No | Disnea | III | Sí | No | 22 | Sí | 50 | 60 | No | No |
AI: aurícula izquierda; DAI: desfibrilador automático implantable; ECG: electrocardiograma; FA: fibrilación auricular; FEVI: fracción de eyección del ventrículo izquierdo; GMM: grosor miocárdico máximo; HFMS: historia familiar de muerte súbita; M: mujer; MCD: miocardiopatía dilatada; MCH: miocardiopatía hipertrófica; MCNC: miocardiopatía no compactada; MP: marcapasos; MSC: muerte súbita cardiaca; NYHA: New York Heart Association; RTG: realce tardío de gadolinio; TRC: terapia de resincronización cardiaca; TSVI: tracto de salida del ventrículo izquierdo; TV: taquicardia ventricular sostenida o no sostenida; V: varón.
Características clínicas, ecocardiográficas y pronóstico de los pacientes afectados de miocardiopatía según la mutación hallada en el gen TNNT2
| Arg92Gln (n=25) | Arg278Cys (n=6) | Arg286His (n=5) | Arg92Trp (n=3) | Arg94His (n=3) | |
|---|---|---|---|---|---|
| Edad (años) | 46,4±17 | 52±14 | 44±24 | 42±23 | 30±13 |
| Varones, n (%) | 14 (56) | 5 (83) | 3 (60) | 1 (33) | 2 (66,6) |
| Fenotipo, n (%) | MCH, 15 (60); MCD, 9 (36); MCNC, 1 (4) | MCH | MCH | MCH | MCH |
| Edad al diagnóstico (años) | 37±16 | 41±14 | 40.6±24 | 35±19 | 23±10 |
| HFMS (%) | 100 (n=21) | 0 (n=3) | 20 (n=5) | 100 (n=1) | 0 (n=1) |
| Presentación (%) | Cribado familiar, 44; disnea, 28; MSC, 20 | Cribado familiar, 33; ECG rutina, 33; disnea, 16; dolor torácico, 16 | ECG rutina, 80; disnea, 20 | Cribado familiar, 100 | Cribado familiar, 33; síncope, 33 |
| Promedio de la NYHA | 1,76 | 1,2 | 1,2 | 1,3 | 1,5 |
| FA, n (%) | (40) | 0 | 0 | 0 | 0 |
| TV, n (%) | (40) | 1 (16,6) | 0 | 0 | 0 |
| GMM (mm) | 15,8±4 | 24±7 | 21±0 | 17,7±6 | |
| Obstrucción TSVI, n (%) | 0 | 1 (16,6) | 1 (25) | 0 | 0 |
| Diámetro AI (mm) | 45±9,6 | 41±9 | 36±10 | 40,6±1,6 | |
| FEVI (%) | 49±15 | 64±5 | 65±4 | 57±21 | 52±17 |
| RTG, n (%) | 10 (91) | 3 (100) | |||
| DAI, n (%) | 11 (44) | 1 (16,6) | 0 | 1 (33) | 1 (33) |
| Muerte, n (%) | 5 (20) | 0 | 0 | 0 | 1 (33) |
AI: aurícula izquierda; DAI: desfibrilador automático implantable; ECG: electrocardiograma; FA: fibrilación auricular; FEVI: fracción de eyección del ventrículo izquierdo; GMM: grosor máximo miocárdico; HFMS: historia familiar de muerte súbita; MCD: miocardiopatía dilatada; MCH: miocardiopatía hipertrófica; MCNC: miocardiopatía no compactada; MSC: muerte súbita cardiaca; NYHA: New York Heart Association; RTG: realce tardío de gadolinio; TSVI: tracto de salida del ventrículo izquierdo; TV: taquicardia ventricular sostenida o no sostenida.
En el análisis de los 57 afectados de miocardiopatía, la media de edad a la presentación fue 37±17 años; 30 (56%) eran varones. Los síntomas iniciales fueron: disnea en 10 (24%), MSC en 5 (12%), dolor torácico en 2 (5%) y síncope en 1 (2%); los demás estaban asintomáticos y el diagnóstico se realizó tras cribado familiar en 17 (40%) o electrocardiograma de rutina en 7 (17%). Las complicaciones durante el seguimiento fueron: taquicardia ventricular o fibrilación ventricular en 3 (7%), insuficiencia cardiaca en 17 (40%), síncope en 6 (14%), dolor torácico en 5 (12%), accidente cerebrovascular en 3 (7%) y fibrilación auricular en 11 (24%).
El GMM medio fue de 18,4±6 (8-35) mm. Los pacientes con mutación Arg92Gln mostraron un GMM medio de 15,8±4mm. Solo 3 (7%) tenían obstrucción del tracto de salida ventricular izquierdo > 30mmHg (un paciente de cada una de las mutaciones Arg278Cys, Arg286His e Ile221Thr). Hubo alta prevalencia de disfunción sistólica ventricular izquierda, en 12 pacientes (31%): 10 con la mutación Arg92Gln, 1 con Arg92Trp y 1 con Arg94His. El diámetro medio de la aurícula izquierda fue 43±9mm. Se realizó cardiorresonancia a 17 pacientes, de los que 15 (88%) presentaron un extenso realce tardío tras gadolinio.
El seguimiento clínico pudo realizarse en todos los pacientes (media, 5±2,5 años). No se observaron cambios significativos en las dimensiones cardiacas o la función sistólica, independientemente del fenotipo cardiaco (MCH, MCD o no compactada).
A 14 individuos (33%) se les implantó un DAI: 8 probandos (6 en prevención primaria y 2 en prevención secundaria) y 6 familiares (5 en prevención primaria y 1 en secundaria). De los 14 pacientes con DAI, 11 tenían la mutación Arg92Gln; 1, Arg92Trp; 1, Arg278Cys (doble mutación en MYBPC3), y 1, Arg94His.
Además, 3 pacientes con Arg92Gln precisaron un dispositivo biventricular para la insuficiencia cardiaca congestiva y 2, un marcapasos bicameral por disfunción sinusal o bloqueo auriculoventricular. Ningún paciente precisó miectomía o trasplante cardiaco.
Durante el seguimiento fallecieron 3 pacientes, uno de insuficiencia cardiaca a los 60 años (paciente con dilatación y disfunción ventricular izquierda grave, Arg92Gln), otro de accidente cerebrovascular (Arg94His) y el tercero de causa desconocida (paciente con insuficiencia cardiaca crónica y disfunción ventricular, fenotipo y familiares con la mutación Arg92Gln, pero sin confirmación genética).
Tres pacientes tuvieron al menos una descarga apropiada del DAI: 2 con Arg92Gln y 1 con Arg278Cys (este con doble mutación en MYBPC3).
En cuanto a las 11 muertes relacionadas con mutaciones en TNNT2 —suma de MSC del análisis del pedigrí (n=6), la primera presentación de la enfermedad (n=3), y las muertes durante el seguimiento (n=2)—, 6 (54,5%) estaban relacionadas con el deporte, 9 (81,8%) eran varones y la media de edad era 21,7±10,9; 7 (63,6%) tenían fenotipo de MCH, mientras que 2 (18,2%) tenían MCD y 2 (18,2%), fenotipo desconocido. El GMM medio era 14,6±5mm; 2 de ellos tuvieron algún episodio de fibrilación auricular. El grupo con Arg92Gln tenía historia familiar de MSC del 100% frente al 28,6% del grupo con mutaciones de TNNT2 distintas de Arg92Gln (p=0,008). La MSC como primer síntoma de la enfermedad ocurrió en 6 pacientes del grupo con Arg92Gln y ninguno en el grupo sin Arg92Gln. Además, se documentaron 3 MSC recuperadas en el grupo con Arg92Gln y ninguna en el otro grupo. En cuanto a las terapias de DAI apropiadas, se produjeron en 3 pacientes del grupo con Arg92Gln y 1 en el otro. Fallecieron durante el seguimiento 2 pacientes del grupo Arg92Gln y ninguno relacionado con otras mutaciones.
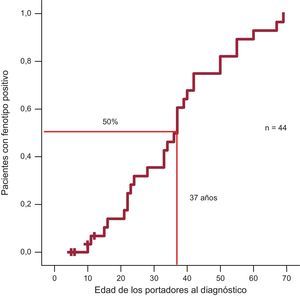
Respecto a la penetrancia, se detectó que un 50% de las personas del grupo Arg92Gln afectadas tenían 37 años de edad (figura 2).

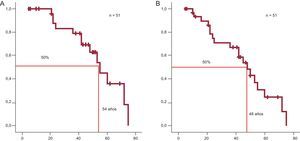
La supervivencia se calculó desde el nacimiento para los portadores de mutaciones en TNNT2, incluidos familiares identificados a partir del árbol familiar que tenían MCH o eran portadores obligados. La mutación Arg92Gln se asoció con mayor tasa de MSC a edad juvenil. La supervivencia media de estos pacientes fue 54 (intervalo de confianza del 95%, 46-62) años, pero si se incluye a los pacientes con MSC recuperada o choque apropiado del DAI, se reduce a 48 años (figura 3). En el grupo con Arg92Gln, la supervivencia a los 55 años fue solo del 50% (intervalo de confianza del 95%, 41-58%).
. B: igual que A, pero incluyendo la muerte súbita cardiaca recuperada y a los pacientes con terapias apropiadas del desfibrilador automático implantable.")
A: supervivencia libre de muerte súbita cardiaca de todos los individuos portadores de la mutación Arg92Gln (incluye a los familiares que tenían miocardiopatía hipertrófica o eran portadores obligados). B: igual que A, pero incluyendo la muerte súbita cardiaca recuperada y a los pacientes con terapias apropiadas del desfibrilador automático implantable.
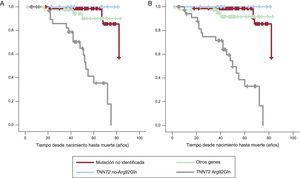
La baja tasa de MSC dificultó el análisis de subgrupos; sin embargo, al comparar probandos con familiares, no hubo diferencias en la mortalidad cardiaca (log rank test de Kaplan-Meier, p=0,62). Se compararon las curvas de supervivencia de la población de estudio: por un lado, la mutación Arg92Gln y, por otro, las otras cinco (debido al diferente perfil pronóstico comentado previamente), otras mutaciones genéticas del sarcómero y pacientes sin mutación identificada (figura 4). Del análisis de las curvas de supervivencia, se concluye que los pacientes con la mutación Arg92Gln tenían una tasa de supervivencia mucho más baja que los otros grupos (otras mutaciones en TNNT2, otros genes sarcoméricos y pacientes con mutación no identificada). Hubo diferencias estadísticamente significativas con el grupo de otras mutaciones en TNNT2 (log rank test, 11,71; p=0,0006). Además, el grupo de pacientes con mutación en TNNT2 en general tuvo una tasa de supervivencia peor que el grupo de otros genes o mutación no identificada, pero esto fue a expensas de los pacientes con la mutación Arg92Gln. No se analizó a los pacientes con dobles mutaciones conjuntamente con otras mutaciones individuales.

A: supervivencia libre de muerte súbita cardiaca en los cuatro grupos de pacientes con miocardiopatía hipertrófica según resultado genético. B: supervivencia libre de muerte súbita cardiaca incluyendo las muertes súbitas cardiacas recuperadas y los pacientes con terapias del desfibrilador automático implantable apropiadas.
Las mutaciones en TNNT2 se consideran una causa infrecuente de MCH (5%)8,9,20,21. En este estudio, la prevalencia fue mayor que en series previas9,10,17,18,22–24, en parte debido a un probable efecto fundador. Este es el estudio con el mayor número de familias y uno de los estudios de más amplia cohorte longitudinal de mutaciones en TNNT2 (tabla 3) desde la publicación del primer estudio que indicaba una alta incidencia de MSC en estos pacientes9. Estos resultados confirman que la MSC en jóvenes es común con algunas mutaciones de este gen, pero esto no se puede extrapolar a todas las mutaciones conocidas.
Estudios publicados sobre supervivencia con mutaciones en el gen TNNT2
| Estudios | N.° de familias | N.° de pacientes | N.° de muertes cardiacas | N.° de muertes súbitas | Mutación |
|---|---|---|---|---|---|
| Watkins et al9 | 11 | 112 | 50 | 39 | Ile79AsnArg92GlnPhe110IleΔGlu160Glu163LysGlu244AspIntron 15 G>AArg278Cys |
| Nakajima-Tanaguchi et al22 | 1 | 4 | 2 | 2 | Ala104Val |
| Moolman et al10 | 2 | 22 | 7 | 7 | Arg92Trp |
| Anan et al17 | 6 | 18 | 2 | 2 | Phe110Ile |
| Torricelli et al18 | 5 | 10 | 0 | 0 | Phe110IleArg130CysΔGlu160Arg92GlnArg278Cys |
| Pasquale et al23 | 20 | 92 | ¿? | 7 | Arg278CysArg92LeuArg92TrpΔGlu163IVS15+1G>AAla104Val Arg278HisArg92Gln Arg94LeuGlu163LysGlu83LysIle79Asn |
| Ripoll-Vera et al, 2015* | 21 | 54 | 11 | 6 | Arg92GlnArg92TrpArg286HisArg278CysArg94HisIle221Thr |
El electrocardiograma es casi siempre anormal, a veces con anormalidades solo leves en el ecocardiograma, lo que resalta el papel de ambas pruebas en la detección de esta enfermedad, algo ya reflejado previamente, pero que es importante destacar.
Hay una diferencia respecto a estudios anteriores9,10,17,18,22–24 en la menor prevalencia de obstrucción del tracto de salida del ventrículo izquierdo en reposo: solo el 7% de los pacientes, algo similar al último estudio publicado23.
Se han identificado seis estudios sobre mutaciones en TNNT2 que analizan la supervivencia9,10,17,18,22,23. Comprenden en total a 258 portadores, 68 fallecidos aparentemente de causas cardiovasculares, la mayoría súbitamente, pero 50 de ellos son de una sola publicación. Por lo tanto, la mayoría de los estudios tienen un bajo número de MSC, lo que concuerda con los datos prospectivos del presente estudio (tabla 3).
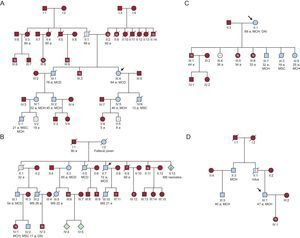
El análisis del árbol familiar (figura 5) muestra una alta prevalencia de MSC en las familias afectadas, pero solo con algunas mutaciones (Arg92Gln principalmente, pero también Arg92Trp y Arg94His). La tasa de MSC fue demasiado pequeña para realizar un adecuado análisis estadístico de subgrupos, al igual que en estudios previos23. El diferente pronóstico en familias con las mismas mutaciones indica que otros mecanismos (genéticos, epigenéticos o factores ambientales) pueden haber tenido alguna influencia.
, 4-Arg92Gln- (B), 11-Arg92Trp- (C) y 12-Arg278Cys- (D). a: años; DAI: desfibrilador automático implantable; MCD: miocardiopatía dilatada; MCH: miocardiopatía hipertrófica; MS: muerte súbita; MSC: muerte súbita cardiaca.")
Algunos estudios previos24 ya documentaron casos de MSC con poca o incluso nula hipertrofia en portadores de mutaciones en TNNT2. También se ha podido observar en algunos pacientes, pero solo con la mutación Arg92Gln. Los pacientes que presentaron MSC tenían más MCH que MCD, y el GMM era de solo 14,6±6mm. La relativa escasez de eventos en la presente cohorte conlleva que no se pueda determinar el riesgo relativo de MSC de los portadores de mutaciones con ecocardiogramas normales.
Este estudio ha detectado importantes diferencias en el pronóstico entre una mutación (Arg92Gln) y las otras cinco mutaciones en TNNT2, los pacientes con mutaciones en otros genes sarcoméricos y también en pacientes con mutación no identificada. Los pacientes con la mutación Arg92Gln tuvieron una presentación más precoz, peor pronóstico, alta incidencia de MSC, fenotipo mixto (MCH con hipertrofia leve, MCD con disfunción ventricular, miocardiopatía no compactada), ausencia de obstrucción, importante fibrosis y frecuente necesidad de DAI o terapia de resincronización.
El adelgazamiento progresivo del miocardio y el deterioro de su función contráctil son un fenómeno bien conocido en la MCH3,4. En este estudio, 9 pacientes tenían ya dilatación ventricular a la presentación (edad, 50,6±14,6 años, todos con Arg92Gln) y 2 fallecieron de insuficiencia cardiaca durante el seguimiento, lo que indica que la progresión a insuficiencia cardiaca puede ser relativamente común en estos pacientes. De hecho, los pacientes con MCH eran más jóvenes (28,8±11,8 años) que los pacientes con MCD. Sin embargo, no se observó ningún caso de progresión a dilatación ventricular, lo que quizá pueda deberse al corto tiempo de seguimiento, por lo que no se puede concluir con total seguridad que los sujetos que presentaban MCD al diagnóstico en realidad eran sujetos con MCH en fase de burn-out.
Un probando con Arg92Gln presentó miocardiopatía no compactada, asociación no descrita hasta ahora. Es importante recordar que, en el caso de pacientes con MCH y MCD, siempre se debe incluir en el estudio genético el gen TNNT2 y posiblemente también, a raíz de estos hallazgos, en casos de miocardiopatía no compactada.
La mutación Arg92Trp afecta al mismo aminoácido que Arg92Gln, por lo que el comportamiento es similar. Se asocia con MCH, hipertrofia leve y alta incidencia de MSC5,19.
La mutación Arg278Cys también se asocia con MCH de presentación tardía e hipertrofia leve-moderada9,18,25–27. La MSC no es frecuente en jóvenes, sino a edades avanzadas. Se asocia con frecuencia con otras variantes patógenas27. En 3 familias, 2 probandos tuvieron mutaciones dobles. No había antecedentes de MSC y el GMM era muy variable (14-35mm).
La mutación Arg286His se ha asociado con MCH19. Las 3 familias tenían MCH con un GMM de 21mm, sin alto riesgo de MSC.
La mutación Arg94His también es una causa conocida de MCH. Los primeros síntomas pueden manifestarse en la infancia y también pueden tener eventos arrítmicos graves en casos con fenotipo aparentemente leve28.
Por último, la mutación Ile221Thr no se había publicado previamente. Se la considera una variante genética rara que afecta a una región funcional relevante. Solo se pudo estudiar el caso índice (que tiene una clara MCH), por lo que no fue posible un estudio de la segregación en la familia para confirmar su patogenicidad.
Se considera que las dobles mutaciones pueden conllevar peor pronóstico. Otros estudios realizados en mutaciones del gen TNNT2 no dieron datos sobre mutaciones adicionales en otros genes. En esta serie se hallaron mutaciones dobles en 4 probandos (19%), pero en ningún familiar estudiado. En la literatura se hallan mutaciones dobles solo en el 5% de las MCH7,23. Solo 1 de los 4 pacientes con doble mutación tuvo una estratificación de riesgo desfavorable, y se le implantó un DAI, pero no hay datos suficientes para establecer un pronóstico relacionado con mutaciones dobles.
Se ha demostrado un probable efecto fundador de la mutación Arg92Gln. De las 10 familias, 9 proceden del mismo pueblo y, mediante estudio genealógico extenso (en archivos parroquiales y censo poblacional), se ha conseguido entroncar 6 de ellas con un antepasado común nacido en 1784. No se ha realizado estudio de haplotipos por no estar disponible en nuestro ámbito por el momento.
LimitacionesAl igual que en estudios previos, pudo haber sesgo de selección debido a que se llevó a cabo en una unidad especializada. Se incluyó a pacientes con confirmación genética y clínica (o patológica), pero también a familiares con fenotipo positivo y a los fallecidos súbitamente, a los que se presumió afectados también pese a no tener confirmación genética.
Las diferencias fenotípicas encontradas entre algunos pacientes con las mismas mutaciones indican que el pronóstico de estos individuos está influido por muchos otros factores, además de la propia mutación.
CONCLUSIONESLa investigación de la correlación genotipo-fenotipo en la MCH sigue siendo un reto. Las mutaciones en el gen TNNT2 fueron más frecuentes en esta serie, en parte por un probable efecto fundador. El perfil clínico y pronóstico dependió en buena medida de la mutación. Los portadores de Arg92Gln tenían un perfil de riesgo significativamente peor que otros pacientes. La MSC fue una complicación frecuente y puede ocurrir en individuos jóvenes con poca o ninguna hipertrofia. La MCD con disfunción ventricular era bastante común entre los portadores de algunas mutaciones (Arg92Gln).
En conjunto, estos hallazgos tienen importantes implicaciones para el estudio clínico y genético de las familias con miocardiopatía, sobre todo el hallazgo de la mutación Arg92Gln, que debería provocar un cambio en el manejo del individuo en cuanto a prevención de la MSC, dada su demostrada malignidad.
FINANCIACIÓNRed de Investigación Cardiovascular del Instituto de Salud Carlos III (RD12/004/0069) y CIBEROBN (Centro de Investigación Biomédica en Red de la Fisiopatología de la Obesidad y la Nutrición) (CB12/03/30038), Madrid, España.
CONFLICTO DE INTERESESNinguno.