Palabras clave
INTRODUCCIÓN
La hipertensión pulmonar (HP) se define1 comoun aumento en la presión arterial pulmonar (PAP)media > 25 mmHg en reposo calculada por el cateterismo cardiaco derecho (CCD). Actualmente no se conoce con exactitud2 el comportamiento normalde la presión pulmonar en el ejercicio, que muestrauna amplia variabilidad según la edad y el grado deentrenamiento físico en el individuo sano. Por lotanto, no es posible establecer una definición de HPen el ejercicio.
CLASIFICACIÓN CLÍNICA DE LA HIPERTENSIÓN PULMONAR
La HP puede presentarse en distintos procesosclínicos3,4 distribuidos en cinco grupos: grupo 1, hipertensión arterial pulmonar (HAP); grupo 2: HPasociada a enfermedad cardiaca izquierda (HPCI);grupo 3: HP asociada a enfermedad respiratoria y/oa hipoxemia; grupo 4: HP tromboembólica crónica (HPTC), y grupo 5: HP por mecanismos pococlaros o multifactoriales.
Esta clasificación1 (tabla 1) está basada en datosclínicos, y en ella se reúnen los procesos y las enfermedades en diferentes grupos que comparten mecanismos fisiopatológicos, presentación clínica y opciones terapéuticas. Respecto de anteriores clasificaciones, ésta incorpora modificaciones sustanciales en el grupo 1. El término HAP familiar sesustituye por HAP heredable, porque se han identificado mutaciones genéticas específicas en casos esporádicos sin antecedentes familiares. Entre lasformas heredables de la HAP se encuentran la HAPidiopática esporádica (HAPI) con mutaciones delínea germinal y casos clínicos con antecedentes familiares con o sin mutaciones identificadas. Estanueva categoría de HAP heredable no exige la realización de pruebas genéticas, puesto que no cambiaría su manejo clínico.

Se ha actualizado la clasificación de las cardiopatías congénitas causantes de HAP para incluir unaversión clínica (síndrome de Eisenmenger, HP asociada a shunt sistemicopulmonares, HP asociada adefectos restrictivos y HP tras la reparación del shunt) y otra anatomofisiopatológica (tabla 2), conel fin de llegar a definir mejor a cada paciente.

Continúa siendo difícil clasificar los trastornos dela enfermedad venooclusiva pulmonar y la hemangiomatosis capilar pulmonar del grupo 1', puestoque comparten algunas características con la HAPI,aunque también manifiestan algunas diferencias.Finalmente, se decidió incluirlas en una categoríadistinta pero no completamente separada de la dela HAP, y por ello se las ha denominado grupo clínico 1'.
BIOPATOLOGÍA DE LA HIPERTENSIÓN PULMONAR
La HAP se define clínicamente como un grupo deenfermedades caracterizadas por el aumento progresivo de la resistencia vascular pulmonar que conduce a la insuficiencia ventricular derecha y lamuerte precoz5. El pronóstico está condicionadopor las interacciones fisiopatológicas complejas entre la tasa de progresión (o regresión) de los cambios obstructivos en la microcirculación pulmonary la respuesta del ventrículo derecho (VD) sobrecargado. Los principales factores pronósticos conocidos en esta enfermedad son expresión de la función ventricular derecha (hemodinámicos, clínicos ybioquímicos). El aumento de la poscarga continúasiendo el principal determinante de la insuficienciacardiaca en pacientes con HAP y HPTC, porque sueliminación como consecuencia de un trasplantepulmonar o una trombendarterectomía supone casi invariablemente una rápida recuperación de la función del VD.
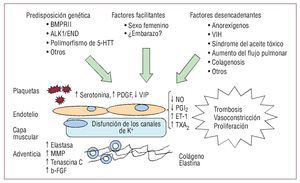
La base fisiopatológica que subyace al aumentode las resistencias vasculares pulmonares es la enfermedad vascular hipertensiva en arterias de pequeño tamaño y arteriolas pulmonares. En su desarrollo participan múltiples factores celulares ymoleculares que dan lugar al remodelado de lapared del vaso por cuatro mecanismos fundamentales, que son la vasoconstricción, la proliferacióncelular, la trombosis y los factores inmunitarios.El origen es desconocido, pero se postula la existencia de una predisposición genética sobre la que deben actuar factores facilitadores y desencadenantes que dan lugar al inicio de la enfermedad6 (fig.1).

Fig. 1. Mecanismos biopatológicos en eldesarrollo de la hipertensión arterial pulmonar. ALK1: activin-receptor-like kinase 1; BMPR II: gen del receptor tipo II de la proteína ósea morfogenética; ENG: endoglina; ET-1: endotelina 1; b-FGF: factor de crecimiento derivado de fibroblastos; 5-HTT:gen del transportador de serotonina; MMP:metaloproteinasas; NO: óxido nítrico; PDGF: factor de crecimiento derivado deplaquetas; PGI2: prostaciclina; TXA2: tromboxano A2; VIH: virus de la inmunodeficiencia humana; VIP: péptido intestinal vasoactivo.
Los diferentes mediadores moleculares implicados en el desarrollo de la enfermedad, su principal mecanismo de actuación y el tipo celular implicado se resumen en la tabla 3. El efecto final deestos mediadores es un desajuste hacia los que favorecen la vasoconstricción, la proliferación celular yla trombosis vascular frente a los que ejercen el mecanismo contrario. El conocimiento de estos mediadores no sólo es importante para entender la historia natural de la enfermedad, sino porque son lasdianas a las que se dirigen los diferentes tratamientos actuales y las líneas de investigación.

Genética de la HAP
En los últimos años7 ha habido grandes avancesen este campo, fundamentalmente en el estudio delos genes BMPR2 (gen del receptor tipo II de laproteína morfogenética ósea), ALK1 (activin-receptor-like kinase 1) y 5HTT (endoglina asociadaa la telangiectasia hemorrágica familiar y el gen deltransportador de serotonina), cuyo polimorfismoLL (dos alelos largos) parece ser más frecuente enpacientes con HAP que en los controles.
Gen BMPR2
Codifica un receptor de membrana pertenecientea la familia de los receptores del factor de crecimiento transformador beta (TGFβ). Se expresa enel endotelio pulmonar, células de músculo liso ymacrófagos, y regula múltiples funciones celulares:proliferación, migración, diferenciación y apoptosis. Su mutación da lugar a haploinsuficiencia, esdecir, una cantidad insuficiente del receptor, lo quefavorece una mayor proliferación celular e inhibición de la apoptosis celular. El gen se localiza en elbrazo largo del cromosoma 2 (2q31,32) y tiene 13exones, y se han descrito hasta 298 mutaciones puntuales diferentes2.
En la HAP «familiar», actualmente clasificadacomo hereditaria, se han descrito mutaciones hastaen el 70% de los casos, con una herencia autosómica dominante. La penetrancia es incompleta, ysólo el 20% de los portadores de la mutación van acontraer la enfermedad. Se da un fenómeno de anticipación genética, es decir, las generaciones posteriores sufren la enfermedad a edades más tempranas. En la HAP idiopática se han descritomutaciones en aproximadamente el 20% de loscasos, en HAP asociada a anorexígenos en el 18%de los casos, en HAP asociada a cardiopatías congénitas en el 6%. En pequeñas series de casos deHAP asociada a colagenosis, virus de la inmunodeficiencia humana (VIH) y aceite tóxico, no se hanencontrado mutaciones en el gen BMPR27-9.
Los pacientes que tienen mutado el gen presentanalgunas diferencias respecto a quienes no lo tienen:la enfermedad aparece a edades más tempranas,presentan un perfil hemodinámico de peor pronóstico y responden menos al test vasodilatador agudo;sin embargo, no existen diferencias en cuanto a lasupervivencia y las características clínicas al diagnóstico10.
EPIDEMIOLOGÍA DE LA HIPERTENSIÓN PULMONAR
En la actualidad no disponemos de datos epidemiológicos comparativos sobre la prevalencia de los diferentes grupos de HP. En un estudio realizado enun laboratorio de ecocardiografía11, la prevalenciade la HP (definida por una PAP sistólica > 40mmHg) entre 4.579 pacientes fue del 10,5%. De los483 casos con HP, el 78,7% padecía cardiopatía izquierda (grupo 2), el 9,7% sufría de enfermedadespulmonares e hipoxemia (grupo 3), el 4,2% teníaHAP (grupo 1) y el 0,6%, HPTC (grupo 4) y fue imposible definir el diagnóstico del 6,8% restante.
Grupo 1
Recientemente se han realizado cuatro registrosnacionales12-15 que han descrito la epidemiología de la HAP. La estimación más baja de la prevalenciade la HAP y la HAPI es de 15 y 5,6 casos/millón, respectivamente, y la más alta es de 26 y 9 casos/millón. La estimación más baja de la incidencia de laHAP es de 2,4 casos/millón/año y la más alta de 7,6.La proporción mujeres:varones está próxima a 2, yla media de edad en el momento del diagnóstico esaproximadamente 50 años, con un número creciente de pacientes mayores de 70 años (un 10-17%según los registros). En el Registro Español deHipertensión Pulmonar (REHAP)15, el 34% de los pacientes tenía HAPI y el 3% tenía antecedentes familiares de HAP. En el subgrupo de la HAP asociada, el 16% padecía enfermedades del tejido conectivo (sobre todo esclerosis sistémica), el 17,5%sufría una cardiopatía congénita, el 6,4% tenía hipertensión portal y el 5,9% estaba infectado por elVIH.
Se cree que la HAP es mucho más prevalente enlos países emergentes16, donde enfermedades relativamente comunes como la esquistosiomasis, laanemia de células falciformes, la infección por VIHy las cardiopatías congénitas pueden complicar suevolución con la aparición de HAP.
Grupo 2
La HPCI es la causa más frecuente de HP. La insuficiencia cardiaca es una enfermedad grave ycomún en los países occidentales, y su incidencia enmayores de 65 años es de aproximadamente10/1.000 personas/año17. En el 44% de los casos, lafracción de eyección del ventrículo izquierdo(FEVI) es normal, la insuficiencia cardiaca se produce por disfunción diastólica y se acompaña deHP hasta en un 83% de los pacientes, según losdatos de un amplio estudio poblacional recientemente publicado18. En el 45% de los pacientes, la insuficiencia cardiaca se produce por disfunción sistólica, y la HP aparece en su evolución en el 60% delos casos.
Grupo 3
Es el de la HP causada por enfermedades pulmonares y/o hipoxemia. En una enfermedad pulmonarobstructiva crónica avanzada, la HP es muy prevalente (> 50%), aunque en general es sólo de gradomoderado19. En la enfermedad pulmonar intersticial, la prevalencia de HP es de un 32-39%. La combinación de fibrosis pulmonar y enfisema conllevauna prevalencia más alta de HP20.
Grupo 4
La incidencia de la HPTC tras una embolia depulmón no es bien conocida, aunque la mayoría delos expertos creen que se produce en un 0,5-2% delos casos1,19. En aproximadamente un 40-50% de los pacientes con HPTC no se objetiva ningún eventoclínico compatible con trombosis venosa profundao embolia pulmonar.
El REHAP15 es el único registro poblacional queincluye HPTC. En él, la incidencia es de 0,9 casos/millón/año y la prevalencia es de 3,2 casos/millón.La HPTC supone el 15% de los pacientes con HPincluidos en este registro.
DIAGNÓSTICO DE LA HIPERTENSIÓN PULMONAR
El diagnóstico de la HP es un proceso escalonadoque parte de la sospecha clínica, requiere confirmación del diagnóstico21, identifica la etiología específica (la HAPI ha de considerarse un diagnóstico deexclusión) y culmina con la evaluación de la gravedad (mediante parámetros clínicos, ecocardiográficos, hemodinámicos, biomarcadores y de capacidad de ejercicio), aspecto clave en la elección deltratamiento y en el seguimiento de los pacientes.
El algoritmo diagnóstico (fig. 2) comienza con laidentificación de los grupos clínicos de HP1 más comunes (grupo 2, cardiopatía izquierda; grupo 3, enfermedades pulmonares), luego distingue el grupo 4(HPTC) y finalmente realiza el diagnóstico y reconoce los diferentes tipos en los grupos 1 (HAP) y 5(miscelánea).

Fig. 2. Algoritmo diagnóstico de la hipertensión pulmonar. ANA: anticuerpos antinucleares; CCD: cateterismo cardiaco derecho; ECO: ecocardiografía transtorácica; ETE: ecocardiograma transesofágico; EVOP: enfermedad venooclusiva pulmonar; HAP: hipertensión arterial pulmonar; HCP: hemangiomatosis capilarpulmonar; HP: hipertensión pulmonar; PAP: presión pulmonar media; PEP: presión de enclavamiento pulmonar; PFP: prueba de función pulmonar; RM: resonancia magnética; TCAR: tomografía computarizada de alta resolución; V/Q: ventilación/perfusión; VIH: virus de la inmunodeficiencia humana.
La sospecha de HP es eminentemente clínica, y sefundamenta en la sintomatología, la presencia defactores de riesgo, los hallazgos de la exploración física y los resultados de exámenes simples como la radiografía de tórax y el ECG. Si la valoración inicialconfirma la sospecha de HP, se realizará un ecocardiograma transtorácico, pruebas de función pulmonar y tomografía computarizada de alta resolución torácica para identificar enfermedadespulmonares (grupo 3) o cardiopatía izquierda (grupo 2). Si no hay datos de enfermedad cardiaca o respiratoria o la HP parece «desproporcionada» para lagravedad de la enfermedad subyacente, se recomienda realizar una gammagrafía pulmonar de ventilación/perfusión (V/Q). Si la gammagrafía V/Qmuestra múltiples defectos de perfusión segmentaria,debe sospecharse HPTC. El diagnóstico final deHPTC requiere una angiografía pulmonar por tomografía computarizada (TC), un CCD y una angiografía pulmonar selectiva. Si se descarta esta posibilidad, una vez confirmado el diagnóstico de HP conel CCD, se estudian los diferentes tipos de HAP.
Es importante destacar algunos aspectos relevantes del diagnóstico:
Clínica
El síntoma de inicio es la disnea de esfuerzo progresiva. Cuando la disfunción del VD progresa, aparecen la angina o el síncope de esfuerzo por incapacidad del VD para adaptar el gasto cardiacoal ejercicio; sólo en fases avanzadas estos síntomasse producen en reposo. En poblaciones conriesgo1-4 de HAP (cardiopatías congénitas, antecedentes de embolia de pulmón, enfermedades deltejido conectivo [ETC], VIH y exposición a tóxicos relacionados con la HP), la aparición de estos síntomas requiere confirmar la enfermedad con unecocardiograma. Únicamente en la esclerodermia,en el candidato a trasplante hepático y en los familiares de los pacientes con HAP hereditaria sedebe realizar un ecocardiograma en ausencia desíntomas.
Ecocardiografía transtorácica
Se debe realizar siempre que se sospeche HP.Permite estimar la presión pulmonar sistólica (PSP),la función sistólica y diastólica del VI y la afecciónvalvular y detectar la presencia de shunt sistemicopulmonar (se utilizará suero salino agitado).
El cálculo de la PSP se basa en la ecuación simplificada de Bernoulli, en la que PSP = 4 × (velocidad máxima de la regurgitación tricuspídea)2 + presión auricular derecha (PAD). La PAD puedecalcularse con el diámetro y la variación respiratoria de la vena cava inferior, aunque a menudo seasume un valor fijo de 5 o 10 mmHg. Cuando resulta difícil medir la velocidad pico de regurgitacióntricuspídea, se recomienda administrar vía intravenosa suero salino agitado, que potencia de formasignificativa la señal Doppler.
En general, la correlación entre la PSP estimadaen el ecocardiograma y la medida en el CCD esbuena (0,57-0,85). Sin embargo, la PSP estimadaen la ecocardiografía puede sobrestimar el valorhemodinámico con una diferencia > 10 mmHghasta en un 48% de los casos, especialmente si elregistro Doppler es de mala calidad22. Además,hay regurgitación tricuspídea en pacientes con PSP> 35 mmHg en aproximadamente un 80% y la capacidad de obtener un flujo que pueda ser analizado varía según la enfermedad subyacente del paciente. Así, en un estudio de 374 pacientes conenfermedad pulmonar, sólo en el 44% se obtuvoun buen registro que permitiera estimar la PSP23. Las cifras de PSP varían con la edad y el peso delpaciente24. Así, se halla PSP > 40 mmHg en el 6%de los individuos mayores de 50 años y en el 5% delos que tienen un índice de masa corporal de 30. Por todo ello, la HP no puede definirse con precisión por un valor de corte de la PSP según el método Doppler.
En el estudio de los pacientes con sospecha deHP1 siempre deben considerarse otras variables ecocardiográficas; la presencia de dilatación de las cámaras derechas (vena cava inferior, aurícula derecha [AD], VD y arteria pulmonar), el aplanamiento o inversión del septo interventricular haciael VI, la presencia de colapso mesosistólico y de untiempo de aceleración menor de 80 ms del flujo pulmonar refuerzan las posibilidades de que el pacientetenga HP significativa.
En las tablas 4 y 5 se propone la actitud clínicabasada en la probabilidad de diagnóstico de HAPsegún criterios ecocardiográficos, síntomas y factores de riesgo, de acuerdo con las guías europeasde práctica clínica actuales1.


Si en el estudio diagnóstico de la HAP se objetivaun shunt sistemicopulmonar con el suero salino agitado o se lo sospecha por hallazgos clínicos, se recomienda realizar un ecocardiograma transesofágico para precisar el diagnóstico. En algunos casos,será necesario realizar técnicas de imagen complementarias como la resonancia magnética, especialmente si el shunt sistemicopulmonar es extracardiaco o en cardiopatías congénitas complejas25.
Si en el estudio ecocardiográfico realizado se sospecha disfunción diastólica como origen de la HPcon función sistólica del VI preservada, se recomienda realizar un estudio completo de la funcióndiastólica26 por Doppler pulsado y tisular del flujomitral, el anillo mitral y las venas pulmonares.Asimismo es necesario valorar la presencia de dilatación de aurícula izquierda y el grado de hipertrofia del VI.
Gammagrafía pulmonar de ventilación/perfusión
Es el método de elección para descartar HPTC enel estudio sistemático de un paciente con HP. Unagammagrafía de V/Q1,25 de probabilidad normal obaja excluye eficazmente la HPTC con una sensibilidad del 90 al 100% y una especificidad del 94 al100%.
Tomografía computarizada de alta resolución
Se recomienda realizar una TC en el diagnósticoinicial de los pacientes con HP1. La TC ofrece imágenes detalladas del parénquima pulmonar y facilita el diagnóstico preciso de la enfermedad pulmonar intersticial (EPI) y el enfisema. En lospacientes con HAP asociada a ETC y que presentandatos de EPI significativa, la TC permite valorarcuánto contribuyen la enfermedad vascular y la posible fibrosis asociada a la enfermedad inmunitaria.
La TC es imprescindible cuando se sospecha clínicamente enfermedad venooclusiva pulmonar(EVPO)/hemagiomatosis capilar pulmonar27. Son características las opacidades en reloj de arena dedistribución centrolobular (opacidad nodular centrolobular difusa) y las líneas septales subpleuralesengrosadas y adenopatías mediastínicas.
Tomografía computarizada multicorte con contraste (angio-tC pulmonar)
Debe realizarse a los pacientes en que la gamma-grafía de V/Q sea compatible con HPTC.
Cateterismo cardiaco derecho
El CCD es imprescindible para realizar el diagnóstico de la HAP, valorar la gravedad del deterioro hemodinámico y analizar la vasorreactividadde la circulación pulmonar1,4,28. Los procedimientos del CCD tienen bajos índices de morbilidad (1,1%)y mortalidad (0,055%) cuando se llevan a cabo encentros especializados.
Las variables que hay que registrar son: PAP (sistólica, diastólica y media), presión en la aurículaderecha, presión de enclavamiento pulmonar (PEP)y presión del VD. De ser posible, el gasto cardiacodebe medirse por triplicado por termodilución opor el método de Fick (obligatorio cuando hay cortocircuitos sistemicopulmonares). Asimismo deberían determinarse las saturaciones de oxígeno de lavena cava superior, la arteria pulmonar y la sangrearterial sistémica y calcular las resistencias vasculares pulmonares.
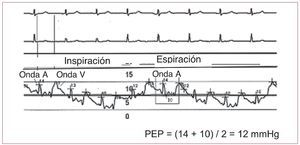
Es necesario ser rigurosos en la determinación dela PEP, ya que es necesario un valor < 15 mmHg para establecer el diagnóstico de HAP. Sin embargo, el método para la medición de la PEP noestá bien estandarizado, y eso produce una ampliavariabilidad entre observadores29. Algunos gruposrealizan la medición al final de la espiración, otrosdurante una pausa de apnea y otros utilizan directamente la medida automática que facilita el programa de ordenador. La influencia de las presionesintratorácicas en las presiones intracardiacas espróxima a cero al final de la espiración, que es elmomento correcto para la medición de la PEP.Además, el catéter debe tener una posición correctaque garantice una buena transmisión de la presiónen aurícula izquierda a través del lecho capilar pulmonar. Se recomienda estudiar las ondas A y V,que deben diferenciarse con claridad, y obtener unaoximetría del extremo distal del catéter para confirmar que la posición es correcta (la saturación deO2 obtenida es similar a la saturación de O2 de lasangre arterial). En ocasiones es necesario penetrarmás distalmente el catéter en el vaso pulmonar paraconseguir un buen trazado.
La onda A se produce simultáneamente a la contracción auricular. En el trazado de la PEP observamos la transmisión de la onda de forma retrógrada y en el ECG aparece atrasada junto al finaldel QRS o inmediatamente después (fig. 3). Se midelos valores máximo y mínimo de la onda A justoantes de la inspiración, y la PEP es la media de esosdos valores. La onda V se produce cuando la sangrellena la aurícula y la válvula mitral se cierra. En eltrazado de la PEP se observa después de la onda Ten el ECG. Una onda V muy prominente puede dificultar la medición exacta de la PEP y suele aparecer por insuficiencia mitral severa o alteracióngrave de la distensibilidad del VI.

Fig. 3. Trazado de presión de enclavamiento pulmonar (PEP) en el que se observan las variaciones respiratorias y la relación temporal de las ondas A y V conel ECG.
Dado el incremento progresivo de la edad de lospacientes con HP y la mayor cantidad de comorbilidades que típicamente se han considerado factoresde riesgo de disfunción diastólica del VI, el diagnóstico diferencial fiable entre HAP y HPCI es cadavez más imprescindible y complicado, a pesar deuna buen técnica en la medición de la PEP19,21. Así, en el Registry to Evaluate Early And Long-termPAH Disease Management (REVEAL)14, que se está realizando en Estados Unidos y en el que se haincluido a 2.525 pacientes (media de edad, 53 ± 14años) con HAP (PEP < 15 mmHg), el 40% tiene hipertensión arterial sistémica; el 33%, obesidad; el21%, apnea del sueño; el 12%, diabetes mellitus, y el4,5%, insuficiencia renal, y se observa una edad másavanzada que en registros previos y una elevadapresencia de factores de riesgo de insuficiencia cardiaca diastólica.
Recientemente se ha realizado un estudio hemodinámico en 3.920 pacientes con HP30, que analiza la fiabilidad de la PEP para discriminar HAP eHPCI comparándola con la presión telediastólicadel VI (patrón de referencia de la precarga del VI).Aproximadamente, la mitad de los pacientes que seclasifican como HAP si se utiliza PEP < 15 mmHg,en realidad tienen HPCI al utilizar el criterio depresión telediastólica del VI < 15 mmHg. A la luzde estos resultados, se propone que, si el pacientepresenta un perfil clínico compatible con HPCI19 (tabla 6), se realice una medición directa de la presión telediastólica del VI si la PEP es < 15 mmHg,para asegurar el diagnóstico de HAP.

En algunos pacientes con un perfil clínico muysospechoso de HPCI que han recibido diuréticos, sepueden observar valores bajos de PEP y de presióntelediastólica del VI. Para verificar el diagnóstico deHAP se recomienda realizar un CCD con sobrecarga de volumen o con ejercicio1,5. Estos procedimientos no están estandarizados y cada laboratoriode hemodinámica tiene su propio protocolo. El cateterismo con sobrecarga de volumen es más sencillo y en esencia consiste en perfundir 1.000 ml desuero fisiológico en 20 min, realizando medicionescada 250 ml. La sobrecarga se detiene cuando laPEP es > 18 mmHg y/o aparecen síntomas.
Test de vasorreactividad pulmonar
Debe realizarse en el momento del diagnósticocon el objetivo de identificar a los pacientes subsidiarios de tratamiento con antagonistas de los canales del calcio. El test debe realizarse con fármacosde acción inmediata, seguros y fáciles de administrar, que generen efectos sistémicos escasos o nulos.Actualmente, el agente más utilizado es el óxido nítrico, aunque hay amplia experiencia con epoprostenol intravenoso y adenosina intravenosa, quetienen mayor riesgo de generar efectos vasodilatadores sitémicos.
Una respuesta aguda positiva1,4,28 (respondedor agudo positivo) se define como una reducción de laPAP media > 10 mmHg para alcanzar un valor absoluto de PAP media < 40 mmHg con un gasto cardiaco invariable o aumentado. Sólo un 10% de lospacientes con HAPI cumplirán estos criterios. Sóloel 50% de los respondedores agudos positivos en laHAPI son respondedores positivos a los antagonistasde los canales del calcio a largo plazo, con prácticanormalización de las presiones pulmonares en el CCD de control. La utilidad del test de vasorreactividad en pacientes con HAP heredable, ETC o VIHno está tan clara. No obstante, la recomendación actual es realizar el test y buscar una respuesta a largoplazo a los antagonistas de los canales del calcio enaquellos en que la prueba sea positiva.
EVALUACIÓN DEL PRONÓSTICO DE LA HAP
La evaluación de la gravedad de los pacientes conHAP tiene lugar entre el proceso diagnóstico y latoma de una decisión terapéutica. La valoración clínica del paciente tiene un papel fundamental en laelección del tratamiento inicial, la evaluación de larespuesta y la posible intensificación de la terapia,si fuera necesario.
Los factores pronósticos31 que se utilizan en laHAP proceden de cohortes de pacientes y puedenno reflejar con exactitud el pronóstico de cada individuo. No hay evidencia científica suficiente parapoder establecer los valores óptimos que alcanzar delos diferentes parámetros, ni tampoco de la relevancia relativa de cada uno de ellos. En la tabla 7 seresumen los factores pronósticos más utilizados1,4,21,28 y se propone un valor óptimo de cada parámetrocomo esquema inicial de trabajo, que cada unidadde hipertensión pulmonar tendrá que adaptar segúnsu propia experiencia y la disponibilidad de las diferentes exploraciones propuestas.

Perfil clínico
La edad, las comorbilidades y la etiología de laHAP, junto con la presencia de insuficiencia cardiaca y la velocidad de progresión, definen el perfilde agresividad de la enfermedad. La hemoptisis ylas arritmias auriculares (fibrilación y aleteo auriculares) son típicas de la HAP severa e indican malpronóstico.
Capacidad de ejercicio
Es uno de los parámetros más clásicos y robustos32. La prueba de marcha de 6 minutos (PM6M)constituye la piedra angular, pero conforme el número de pacientes en clase funcional II y con laPM6M > 400 m se incrementa, es necesaria la incorporación de herramientas más finas para valorar loscambios en la capacidad de ejercicio. La ergoespirometría es la de elección. La información sobre elvalor pronóstico de la ergoespirometría es escasa enel momento actual, pero se perfila como un campocon importante desarrollo en un futuro inmediato33.
Biomarcadores
NT-proBNP y BNP son los más ampliamenteutilizados, y se consideran excelentes marcadores dela severidad de la disfunción del VD. Sus valores seincrementan con el deterioro progresivo de la capacidad de ejercicio o de la clase funcional, y disminuyen si se produce una respuesta positiva al tratamiento34.
Función de ventrículo derecho
La función del VD es el principal determinantepronóstico en los pacientes con HAP35. El ecocardiograma es una herramienta sencilla y asequible: la dilatación de las cavidades derechas, el índice de excentricidad diastólico del VI, el índice TAPSE36 (desplazamiento sistólico del anillo tricuspídeo haciael ápex), el índice de TEI (índice combinado de función miocárdica) y la presencia de derrame pericárdico nos permiten valorar la adaptación del VD a lahipertensión pulmonar. Es de destacar la nula información pronóstica que aporta la estimación de lapresión sistólica pulmonar en el seguimiento de estospacientes. La resonancia magnética25 es la técnica de imagen que mejor evalúa la función del VD: volumen, grado de hipertrofia y fracción de eyección.El trabajo de estratificación pronóstica de los pacientes con HAP con esta técnica está en sus primeras fases, pero con un indudable potencial de desarrollo. Finalmente, los parámetros hemodinámicosde función del VD han sido ampliamenteutilizados1,4,28 y tienen un claro papel en la valoraciónde la severidad y el pronóstico de la HAP.
VÍA CLÍNICA DEL PACIENTE CON HIPERTENSIÓN PULMONAR
La HP, especialmente la HAP y la HPTC, es unasituación clínica grave. El diagnóstico suele establecerse en fases avanzadas de la enfermedad y en laatención clínica de los pacientes intervienen distintos especialistas pertenecientes a diferentes niveles asistenciales. Asimismo, algunos de los procedimientos diagnósticos y terapéuticos empleadostienen una complejidad elevada y requieren experiencia en su ejecución, por lo que hay consenso enque los pacientes con HAP o HPTC sean derivadosa unidades de referencia1,4,28. Todo ello hace necesaria una vía clínica que permita mejorar los procesos asistenciales relacionados con el paciente.
Habitualmente el diagnóstico de sospecha de HPlo establecen especialistas del centro más próximoal paciente (unidad clínica local). Si tras los exámenes iniciales se cree que el paciente puede sufrirHAP, HPTC o HP de origen multifactorial, se debederivarlo a una unidad de referencia en HP pararealizar el CCD (tabla 8). No es aconsejable realizarel CCD antes de la derivación, excepto en casos enque así se haya acordado con la unidad dereferencia1,28. Los pacientes con enfermedad cardiaca o respiratoria en que se sospecha HP desproporcionada también deben ser derivados a unaunidad de referencia en HP.

Los pacientes con HP pueden deteriorarse rápidamente28, por lo que es importante establecer unosplazos breves para completar el estudio diagnósticoe iniciar el tratamiento, y es imprescindible la coordinación fluida entre la unidad clínica local y elcentro de referencia (tabla 9).

Para los pacientes que residen lejos de la unidadde referencia, es aconsejable que también sean atendidos por un especialista de un centro hospitalariocercano, preferiblemente con interés en HP, que actuaría como unidad clínica local, efectuaría el seguimiento más próximo al paciente y lo atendería enprimera instancia en caso de complicaciones. Enestas circunstancias el paciente debería seguir unplan de cuidados compartidos entre la unidad clínica local y la de referencia.
En general, el curso evolutivo de la HP es de deterioro progresivo con episodios intercurrentes dedescompensación. El seguimiento clínico debe mantenerse hasta que se produzca la muerte o se realiceel trasplante. En las fases avanzadas e irreversiblesde la enfermedad, se debe considerar los cuidadospaliativos del paciente de común acuerdo con éste,el entorno familiar, la unidad clínica local y launidad de referencia.
Unidades de referencia
Recientemente, la Sociedad Española deNeumología y Cirugía Torácica, junto con laSociedad Española de Cardiología, en un documento de consenso nacional28, y la Sociedad Europea de Cardiología y la Sociedad RespiratoriaEuropea en las guías clínicas europeas28 han efectuado recomendaciones sobre las prestaciones y elvolumen de actividad de las unidades de referenciaen HP, que se resumen en la tabla 10.

En determinadas circunstancias, los pacientes conHP requieren procedimientos altamente especializados que no forman parte de las atribuciones propias de las unidades de referencia. Estas situacionesse plantean específicamente en la HPTC (endarterectomía pulmonar), las cardiopatías congénitas, laHAP hereditaria y la portopulmonar y en el trasplante pulmonar o cardiopulmonar. Las unidadesde referencia deberán actuar de forma coordinada ytener protocolos de derivación preestablecidos conlas unidades subespecializadas para atender adecuadamente estos casos.
CONCLUSIONES
Recientemente se han producido importantesavances en el conocimiento de la biopatología, eldiagnóstico, la epidemiología y el pronóstico de lospacientes con HAP que han permitido mejorar notablemente la calidad y la eficacia de su atenciónclínica, aunque todavía no se ha conseguido su curación. El diagnóstico correcto y la estimación multifactorial del pronóstico son aspectos clave en esteproceso y requieren experiencia y capacitación técnica para conseguir un resultado óptimo. Por elloes necesario estructurar la organización asistencialy establecer una vía clínica para los pacientes conHAP.
ABREVIATURAS
CCD: cateterismo cardiaco derecho.
HAP: hipertensión arterial pulmonar.
HAPI: hipertensión arterial pulmonar idiopática.
HP: hipertensión pulmonar.
HPTC: hipertensión pulmonar tromboembólicacrónica.
PEP: presión de enclavamiento pulmonar.
VIH: virus de la inmunodeficiencia humana.
Full English text available from: www.revespcardiol.org
La Dra. Pilar Escribano Subias es investigadora de la red de investigacióncooperativa REDINSCOR del Ministerio de Sanidad y Consumo.
Correspondencia:
Dra. P. Escribano Subias.
Avda. Córdoba, s/n. 28041 Madrid. España.
Correo electrónico: pilar.escribano@telefonica.net