HCN4 variants have been reported to cause combined sick sinus syndrome (SSS) and left ventricular noncompaction (LVNC) cardiomyopathy. This relationship has been proven in few cases and no previous patients have associated left atrial dilatation (LAD). Our objective was to study a familial disorder characterized by SSS, LAD, and hypertrabeculation/LVNC and to identify the underlying genetic and electrophysiological characteristics.
MethodsA family with SSS and LVNC underwent a clinical, genetic, and electrophysiological assessment. They were studied via electrocardiography, Holter recording, echocardiography, and exercise stress tests; cardiac magnetic resonance imaging was additionally performed in affected individuals. Genetic testing was undertaken with targeted next-generation sequencing, as well as a functional study of the candidate variant in Chinese hamster ovary cells.
ResultsTwelve members of the family had sinus bradycardia, associated with complete criteria of LVNC in 4 members and hypertrabeculation in 6 others, as well as LAD in 9 members. A HCN4 c.1123C>T;(p.R375C) variant was present in heterozygosis in all affected patients and absent in unaffected individuals. Electrophysiological analyses showed that the amplitude and densities of the HCN4 currents (IHCN4) generated by mutant p.R375C HCN4 channels were significantly lower than those generated by wild-type channels.
ConclusionsThe combined phenotype of SSS, LAD, and LVNC is associated with the heritable HCN4 c.1123C>T;(p.R375C) variant. HCN4 variants should be included in the genetic diagnosis of LVNC cardiomyopathy and of patients with familial forms of SSS, as well as of individuals with sinus bradycardia and LAD.
Keywords
Tetramers of hyperpolarization-activated cyclic nucleotide-gated channel (HCN4) subunits constitute the ion channels that conduct the hyperpolarization-activated “funny” current (If), which plays a critical role in sinoatrial node pacemaker activity. The involvement of HCN4 loss-of-function variants in familial forms of sinus sick syndrome (SSS) was first noted in 2003.1 The clinical course of these syndromes includes supraventricular arrhythmias (particularly atrial fibrillation [AF]), early pacemaker need, cardioembolic complications, and even sudden cardiac death.2,3
More recently, HCN4 variants have been associated with cardiac abnormalities such as left ventricular noncompaction (LVNC), mitral valve prolapses, and ascending aorta dilatation.4–6
We report a large family with inherited SSS associated with left atrial dilatation (LAD) and LVNC caused by the rare p.R375C HCN4 variant, which exhibits complete penetrance and a benign clinical course.
METHODSGenetic testingA genetic study including 173 genes was conducted in the index patient using targeted next-generation sequencing (Illumina HiSeq; Applied Biosystems 3730 DNA Analyzer, Thermo Fisher Inc, United States). Candidate variants were considered according to the usual pathogenicity criteria.7 DNA was obtained from blood samples in adults and from saliva in pediatric patients. Segregation was considered to be present if a variant was found in all affected individuals and if it was absent in individuals without phenotype. Significant cosegregation was defined using a cutoff of N1/32.8
Cell culture and electrophysiological analysis of the candidate variantHCN4 currents (IHCN4) were recorded using the whole-cell patch-clamp technique in Chinese hamster ovary cells transiently transfected with either wild-type (WT) or mutated HCN4 channels, as previously described.9 Current densities were calculated by normalization of the current amplitude to the capacitance of the cell. The voltage-dependence of HCN4 channel activation was analyzed and Boltzmann functions were fitted to the data to calculate the membrane potential at which 50% of the channels were activated (Vh) and the slope (k) of the curves. The ion selectivity of the HCN4 channels was assessed by measuring the IHCN4 density relationships of fully activated channels.
Patients and clinical assessmentThe clinical evaluation and genetic study of all family members conformed to the principles outlined in the Declaration of Helsinki. Each patient was informed about the objectives of the research. In the case of minors, the consent of the parents was always required. All individuals were evaluated in our inherited cardiovascular disease unit. The initial assessment included 12-lead electrocardiography and transthoracic echocardiography.
We considered individuals as being possibly affected if their heart rate (HR) was below 60 beats per minute (bpm) (or according to the age limit reference in pediatric patients10) and/or if they exhibited hypertrabeculation/noncompaction on transthoracic echocardiography according to the standard criteria.11,12 The study was completed with cardiac magnetic resonance (CMR), 24-hour Holter electrocardiography, and cardiopulmonary exercise testing combined with transthoracic echocardiography. LVNC was diagnosed in patients who met Petersen and/or Jacquier criteria.13,14 In pediatric patients, the indications for complementary tests were individualized.
Statistical analysesAll statistical analyses were performed using IBM SPSS version 20.0.0 (IBM Corp, United States). Qualitative variables are expressed as absolute number (n) and percentage (%) and continuous values are reported as mean±standard deviation or median and range.
Results of the functional study are expressed as mean±standard error of the mean and were compared using t test, ANOVA followed by Tukey test, or Mann-Whitney U test, as appropriate, as well as using multilevel mixed-effects models. P <.05 was considered statistically significant for all analyses. More details on the methodology can be found in the .
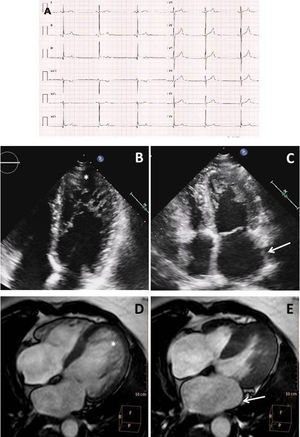
RESULTSClinical assessment in the index patientThe index patient was an 18-year-old man (III.19) with sinus bradycardia (SB) and no relevant medical history. Resting electrocardiography showed narrow QRS and normal PR and QT intervals with a J-point elevation in the inferior leads (figure 1A). Holter recordings demonstrated minimum and average HRs of 32 and 44 bpm, respectively. Exercise testing showed excellent functional class. Transthoracic echocardiography revealed LAD, left ventricular dilatation with hypertrabeculation, and a normal ejection fraction (figure 1B,C). LVNC diagnostic criteria were confirmed by CMR (figure 1D,E).
. C: endsystolic frame showing left atrial dilatation (arrow). D: CMR showing biventricular hypertrabeculation (arrows) meeting LVNC criteria. E: CMR, with the endsystolic phase showing left atrial dilatation (arrow). CMR, cardiac magnetic resonance; LVNC, left ventricular noncompaction.")
Complementary tests in the index patient. A: electrocardiography indicating sinus bradycardia. B: apical 4-chamber transthoracic echocardiography exhibiting deep apical hypertrabeculation in the left ventricle (asterisk). C: endsystolic frame showing left atrial dilatation (arrow). D: CMR showing biventricular hypertrabeculation (arrows) meeting LVNC criteria. E: CMR, with the endsystolic phase showing left atrial dilatation (arrow). CMR, cardiac magnetic resonance; LVNC, left ventricular noncompaction.
There was no family history of pacemaker implantation or sudden cardiac death. All relatives were asymptomatic at the time of the initial evaluation, except the index patient's mother (II.10). She had a history of paroxysmal AF and underwent successful pulmonary vein ablation. The maternal grandmother (I.2) died of stroke at 85 years old and the grandfather (I.1) died of heart failure of unknown origin at 72.
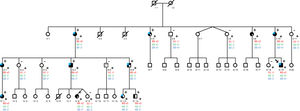
Evaluation of 22 individuals was possible: 2 first-degree relatives (mother and sister) and 20 other relatives in the maternal line (figure 2). In the first evaluation, 12 individuals seemed to be affected: all had SB. This was later confirmed by a positive genetic test.

Pedigree of the family with the HCN4 variant. Numbers below the subject symbol denote the family member's identification. Squares/circles: male/female. Asterisk (*): individuals studied by our clinic team. Slashed symbols: deceased persons. Shaded left: sinus bradycardia. Shaded right: left ventricular noncompaction. Half shaded right: hypertrabeculation without LVNC criteria. Blue shading in the upper left: left atrial dilatation. G1: genotype for HCN4:p.R375C; G2: genotype for FHOD3:p.F567L; G3: genotype for DSP:p.R1544S. +/-: heterozygosis; -/- homozygosis (not mutated). LVNC, left ventricular noncompaction.
Genetic analysis in the index patient identified 3 variants of uncertain significance in heterozygosis: NM_005477.2:c.1123C>T;(p.R375C) in the HCN4 gene, NM_001281740.1:c.1701C>A;(p.F567L) in the FHOD3 gene, and NM_004415.2:c.4632G>T;(p.R1544S) in the DSP gene. Of these variants, only HCN4 c.1123C>T;(p.R375C) segregated with the familial disease. The p.R375C variant was previously annotated (rs755356387) with a total allele frequency of 0.000003977 (Genome Aggregation Database [gnomAD]).15 SB had complete penetrance, even in pediatric individuals. Thus, by considering affected and nonaffected family members, SB showed very strong cosegregation (n = 1/218). LVNC had incomplete penetrance, with variable expressivity among individuals, but also showed significant cosegregation (n=1/25). In this case, only affected individuals were taken into account for cosegregation calculation (figure 2). The main characteristics of the HCN4 variant carriers are shown in table 1 and table 2.
Baseline characteristics and electrocardiogram, exercise test, and echocardiogram data of the p.R375C HCN4 variant carrier's family
| Baseline characteristics | ECG data | Exercise test | Transthoracic echocardiography | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Sex | Age | Rhythm | HR, bpm | Sinus pauses> 3 s or AVB | Pacemaker | NYHA FC | % MHR | Peak V02, mL/kg/min | % of predicted V02 | Hypertrabeculation pattern | Indexed LA volume, mL/m2 |
| III.19a | M | 20 | SR | 35 | No | No | I | 72 | 36.5 | 87 | LVNC | 44.7 |
| II.2b | F | 69 | SR | 50 | No | No | I | 69 | 22.9 | 114 | Hypertrabeculation | 43.9 |
| II.5 | F | 68 | SR | 59 | No | No | I | 87 | 25 | 138 | Hypertrabeculation | 39.9 |
| II.9 | F | 61 | SR | 55 | No | No | - | - | - | - | Hypertrabeculation | 21.3 |
| II.10b | F | 56 | SR | 40 | No | No | I | 67 | 26.3 | 125 | LVNC | 30.3 |
| III.1 | F | 48 | SR | 59 | No | No | I | 99 | 23.1 | 94 | LVNC | 52.4 |
| III.4 | M | 42 | SR | 41 | No | No | I | 83 | 37.8 | 113 | LVNC | 37.4 |
| III.6 | M | 36 | SR | 40 | No | No | I | 89 | 39.4 | 117 | Hypertrabeculation | 39.7 |
| IV.1 | F | 16 | SR | 37 | No | No | I | 87 | 42.5 | 125 | LVNC | 30.0 |
| IV.8 | F | 5 | SR | 75 | No | No | I | 79 | - | - | Hypertrabeculation | 26.6 |
| IV.13 | F | 12 | SR | 50 | No | No | I | 88 | - | - | Normal | 40.8 |
| IV.14 | M | 9 | SR | 55 | No | No | I | 77 | - | - | Normal | 14.4 |
AVB, atrioventricular block; bpm, beats per minute; ECG, electrocardiogram; F, female; HR, heart rate; LA, left atrial; LVNC, left ventricular noncompaction; M, male; MHR, maximum heart rate; NYHA FC, New York Heart Association functional class; SR, sinus rhythm; VO2, oxygen consumption.
Cardiac magnetic resonance data of the p.R375C HCN4 variant carrier's family
| Patient | CMR data | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hypertrabeculation pattern | LVEF, % | RVEF, % | Indexed LVTDV, mL/m2 | Indexed RVTDV, mL/m2 | LGE | GLS-FT, % | GCS-FT, % | Native T1, ms | ECV, % | Indexed LA volume, mL/m2 | |||
| Visual hypertrabeculation | Petersen | Jacquier | |||||||||||
| III.19a | Yes | No | Yes | 59 | 64 | 147 | 134 | No | −27 | −20.39 | 918 | 22.6 | 46.4 |
| II.2b | No | No | No | 65 | 72 | 86 | 81 | No | −25.63 | −20.04 | - | - | 54.1 |
| II.5 | No | No | No | 69 | 73 | 91 | 86 | No | −27.55 | −22.28 | 968 | 20.4 | 60.3 |
| II.9 | - | - | - | - | - | - | - | - | - | - | - | - | |
| II.10b | Yes | No | No | 67 | 80 | 105 | 91 | No | −27.09 | −25.98 | - | - | 51.7 |
| III.1 | Yes | Yes | Yes | 52.3 | 62 | 96 | 75 | No | −17.81 | −16.7 | - | - | 41.3 |
| III.4 | Yes | Yes | Yes | 56.5 | 64 | 128 | 114 | No | −21.97 | −20.01 | 950 | 20.5 | 53.2 |
| III.6 | Yes | No | Yes | 54.5 | 54.9 | 121 | 116 | No | −19.87 | −17.99 | 999 | 21.8 | 39.7 |
| IV.1 | Yes | No | No | 64 | 67 | 119 | 112 | No | −23.06 | −21.96 | 957 | 23.2 | 49.5 |
| IV.8 | - | - | - | - | - | - | - | - | - | - | - | - | - |
| IV.13 | - | - | - | - | - | - | - | - | - | - | - | - | - |
| IV.14 | - | - | - | - | - | - | - | - | - | - | - | - | - |
CMR, cardiac magnetic resonance; ECV, extracellular volume; GCS-FT, global circumferential strain by feature tracking; GLS-FT, global longitudinal strain by feature tracking; LA, left atrial; LGE, late gadolinium enhancement; LVEF, left ventricular ejection fraction; LVTDV, left ventricular telediastolic volume; RVEF, right ventricular ejection fraction; RVTDV, right ventricular telediastolic volume.
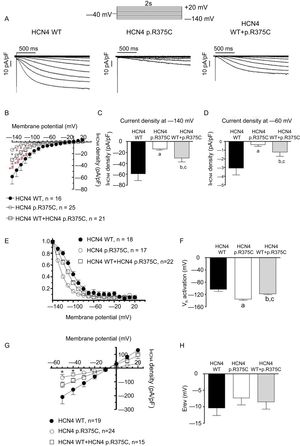
The left panel in figure 3A shows the IHCN4 traces generated by applying 2-second pulses from −140 to +20mV in 10-mV steps from a holding potential of −40mV to a Chinese hamster ovary cell expressing WT HCN4 channels. Hyperpolarizing pulses generated an inward current that slowly activated until reaching a steady-state level whose amplitude progressively decreased at more positive potentials. Compared with WT channels, p.R375C HCN4 channels generated a smaller and much more slowly activating inward current (figure 3A, middle panel). Considering the heterozygous condition of carriers, cells were cotransfected with WT and p.R375C HCN4 channels in a 1:1 ratio. The IHCN4 amplitude generated by the cotransfection was lower than that generated by WT channels (figure 3A, right panel). By analyzing the IHCN4 density at the different membrane potentials, we confirmed that the IHCN4 densities generated by p.R375C channels were significantly lower than the densities generated by WT+p.R375C and WT channels (n ≥ 16; P <.01; figure 3B). Additionally, the IHCN4 decrease was evident both at very negative (−140mV, figure 3C) and physiological (−60mV, figure 3D) membrane potentials (n ≥ 16; P <.01). Interestingly, the current density generated by the cotransfection of WT and p.R375C channels was half of that generated by WT channels (n ≥ 16; P <.01). These results suggest that p.R375C channels do not act as “poisoning proteins” with a dominant negative effect.
 or −60mV (panel D). E: tail current densities generated by different channels by applying 1-second pulses to −140mV were normalized and plotted against the membrane potential of the test pulse. Continuous lines represent the Boltzmann fit to the data. F: the membrane potential activating 50% (Vh) of the WT, p.R375C, and WT+p.R735C HCN4 channels. G: fully activated IHCN4 density–voltage relationships generated by WT, p.R375C, and WT+p.R375C channels. Continuous lines represent the linear regression to the data. H: Erev of the channels calculated from the intersection of the linear regression to the data with the abscissas axis of each individual experiment. In B to H, each point/bar represents the mean±SEM of ≥ 15 experiments/cells from ≥ 3 dishes. CHO, Chinese hamster ovary; Erev, reversal potential; IHCN4, HCN4 currents; WT, wild-type. aP <.01 vs HCN4 WT. bP <.05 vs HCN4 WT. cP <.05 vs p.R375C.")
Electrophysiological analysis of the HCN4 variant. A: families of IHCN4 traces generated in CHO cells transiently expressing HCN4 WT, p.R375C, and WT+p.R375C channels by applying the protocol depicted at the top. B: density–voltage relationships generated in cells expressing WT, p.R375C, and WT+p.R375C channels. C and D: IHCN4 density generated by HCN4 WT, p.R375C, and WT+p.R375C channels with the application of pulses at −140 (panel C) or −60mV (panel D). E: tail current densities generated by different channels by applying 1-second pulses to −140mV were normalized and plotted against the membrane potential of the test pulse. Continuous lines represent the Boltzmann fit to the data. F: the membrane potential activating 50% (Vh) of the WT, p.R375C, and WT+p.R735C HCN4 channels. G: fully activated IHCN4 density–voltage relationships generated by WT, p.R375C, and WT+p.R375C channels. Continuous lines represent the linear regression to the data. H: Erev of the channels calculated from the intersection of the linear regression to the data with the abscissas axis of each individual experiment. In B to H, each point/bar represents the mean±SEM of ≥ 15 experiments/cells from ≥ 3 dishes. CHO, Chinese hamster ovary; Erev, reversal potential; IHCN4, HCN4 currents; WT, wild-type.
aP <.01 vs HCN4 WT.
bP <.05 vs HCN4 WT.
cP <.05 vs p.R375C.
To quantify the activation kinetics, a monoexponential function was fitted to the activation of traces generated by pulses to −130mV. Time constants of the IHCN4 activation (τ) averaged 495±44, 1960±387, and 727±85 milliseconds for WT, p.R375C, and p.R375C+WT HCN4 channels, respectively (n ≥ 16, P <.01).
An analysis of the voltage-dependence of HCN4 channel activation is shown in figure 3E (see : Patch-clamp recordings). p.R375C channels were activated at more negative potentials compared with WT channels, an effect that probably accounts for the reduction in the current density produced by the variant. Indeed, the Vh was significantly hyperpolarized (n ≥ 17; P <.01) (figure 3F). WT+p.R375C channel activation was also shifted to negative potentials compared with WT channels (n ≥ 18; P <.05; figure 3E,F).
To determine whether the variant alters the ion selectivity of HCN4 channels, we measured the IHCN4 density relationships of fully activated channels (figure 3G). The reversal potential (Erev) was calculated from the intersection of the linear regression of the data with the abscissa axis at extracellular and intracellular K+ concentrations of 30 and 142mM, respectively (see : Patch-clamp recordings). The reversal potential was not modified by p.R375C channels alone or by their coexpression with WT channels (n ≥ 15; P> .05) (figure 3G,H). These results suggest that the variant did not modify the ion selectivity of HCN4 channels.
Complementary studies in HCN4 variant carriers and follow-upBecause participant II.9 underwent clinical follow-up in another medical center, we do not have further data on this person. All genetic carriers met the criteria for sinus dysfunction and none showed AF in Holter electrocardiography (table 1). Interestingly, IV:8 and IV:9 were 5-year-old twins, but IV:9 was not a genetic carrier and her resting HR was 100 bpm compared with the 75 bpm of IV:8.
Transthoracic echocardiography revealed the hypertrabeculation/LVNC phenotype in 10 of the 12 genetic carriers (83%), LAD in 7 of the 12 genetic carriers (58%), and a normal heart in 2 pediatric individuals (table 1). No other abnormalities were found in any participants.
CMR was performed in 8 patients (pediatric patients were excluded) (table 2). Four patients met LVNC diagnostic criteria and 4 other patients showed nonpathological ventricular hypertrabeculation. Five patients had mild left ventricular dilatation and normal right ventricular size, and the index patient (III.19) exhibited severe biventricular dilatation. Three HCN4 variant carriers (III.1, III.4, and III.6) had borderline LVEF. Regarding myocardial deformation data, just 1 patient (III.1) had both a pathological global longitudinal strain (GLS) and global circumferential strain (GCS). None of the patients exhibited late gadolinium enhancement and all patients showed normal values of the native T1 (965±15.8 milliseconds) and extracellular volume (21.7%±1.2%). All patients exhibited LAD, with 89% showing severe enlargement, and all had normal atrial ejection fraction on CMR (ejection fraction, 56.6%; GLS, 35.8%).
Cardiopulmonary exercise testing was performed in all adult genetic carriers (table 1). All carriers completed the third stage of the Bruce protocol and 6 developed ventricular ectopic beats. Stress echocardiograms showed a positive contractile reserve without other pathological indicators. All patients had a normal functional class for the theoretical predicted peak VO2 (111%±15%), normal behavior of the O2 pulse (142%±30%), and VO2 at the anaerobic threshold higher than predicted (72%±15%). Pediatric carriers were evaluated by a conventional treadmill exercise test and all had a normal functional class with no significant arrhythmias.
At the end of this study, all family members were alive (mean follow-up, 29±9 months) and none had developed heart failure. Apart from the patient with AF, no other arrhythmias have been documented. Accordingly, no patient has needed a pacemaker or implantable cardioverter-defibrillator.
DISCUSSIONThis study describes a large family with a combined phenotype of SB, LAD, and LVNC due to the c.1123C>T;(p.R375C) variant in the HCN4 gene. This variant was previously annotated (RCV000693215.1; rs755356387) and described in 1 individual with sudden cardiac death and LVNC/SB, but no segregation or functional study was performed.16 According to the consensus recommendation of the American College of Medical Genetics and Genomics,7 it is classified as a “variant of uncertain significance”.
HCN4 variants were initially linked to inherited SSS without structural heart disease.2 In 2014, the association between LVNC and SSS was first described in several families with HCN4 variants.4,5 Only 68 cases belonging to 16 families have thus far been described. The presence of left ventricular hypertrabeculation/LVNC and different degrees of sinus node dysfunction, including tachycardia-bradycardia syndrome with AF,17 has been constant in all cases described, sometimes associated with other disorders such as mitral valve prolapse4,5 or ascending aorta dilatation.6 Pacemaker need seems high in this group of patients and, among all cases described, 4 patients experienced aborted sudden cardiac death due to ventricular fibrillation and 1 patient experienced sudden cardiac death.5,16,18
We believe that the pathogenicity of HCN4 c.1123C>T;(p.R375C) has been proven according to reference criteria.7 Family studies show an autosomal dominant inheritance pattern with complete penetrance for SB and incomplete but equally high penetrance for LAD and noncompaction.8,17 The high penetrance of SB present in pediatric individuals has previously been reported4,17 and could be a marker of the disease. The HCN4 c.1123C>T;(p.R375C) carriers in our family showed a benign course compared with previous work.16 The only complication identified during follow-up was an episode of persistent AF with excellent rhythm control, which was managed using pulmonary vein ablation.
Mechanisms of HCN4 variants underlying SSSOur results have shown that the p.R375C variant profoundly modifies the voltage-dependence and kinetics of HCN4 channel activation. This significantly decreased the channel availability, diminishing the density of the current generated. The causality of HCN4 variants has been demonstrated via cellular electrophysiological analysis.4,5,17–19 Mutated HCN4 channels were nonfunctional, resulting in If current reduction. In the present study, the electrophysiological data provided strong evidence for the causality of the p.R375C variant in the pathogenesis of the observed SB.
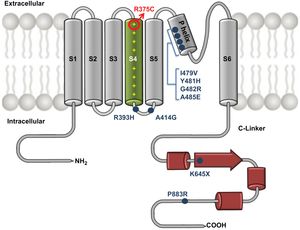
The p.R375C residue is located in the S4 segment of HCN4 channels and is 1 of the 7 positively charged residues of this channel domain (figure 4). Indeed, the S4 segment is the voltage sensor of the channel, and the variant replaces 1 of the positively charged Arg residues with a polar neutral residue (Cys). This perfectly explains the profound changes produced by the variant in the voltage-dependence and kinetics of channel activation. Similar results were previously described for the p.R378C variant found in a patient with SSS.20
 channel schematic. Illustration of the structure of the HCN4 channel depicting variants combining the LVNC and SSS phenotype described thus far. Arg375 (circle) is located in the S4 segment and is 1 of the 7 positively charged residues of this domain that constitutes the channel voltage sensor. LVNC, left ventricular noncompaction; SSS, sinus sick syndrome.")
Mutant HCN4 c.1123C>T;(p.R375C) channel schematic. Illustration of the structure of the HCN4 channel depicting variants combining the LVNC and SSS phenotype described thus far. Arg375 (circle) is located in the S4 segment and is 1 of the 7 positively charged residues of this domain that constitutes the channel voltage sensor. LVNC, left ventricular noncompaction; SSS, sinus sick syndrome.
LVNC cardiomyopathy has been linked to different variants affecting sarcomeric, cytoskeletal, and nuclear membrane genes. Animal models indicate that the hypertrabeculation results from altered regulation of cell proliferation, differentiation, and maturation during the formation of the ventricular wall.21
Thus far, at least 8 different HCN4 variants associated with this combined phenotype have been published (P883R, K645X, A485E, G482R, Y481H, I479V, A414G, R393H).4,5,17–19 There may be a relationship between the development of the cardiac conduction system and the myocardium maturation that explains the concomitant presence of cardiomyopathy and specific arrhythmias.22 These HCN4 variants could be considered the “final common pathway” that leads to a common phenotype,23 although it remains to be determined how the hypertrabeculation develops. Another hypothesis for the occurrence of LVNC is an acquired adaptive remodeling feature in response to SB.5 Previous studies have reported that genes encoding ion channels (SCN5A and RYR2) are involved in cardiomyopathy pathophysiology.24,25 In this study, we found some confusing data regarding whether our family has an acquired phenotypic trait or a true cardiomyopathy. Almost all carriers had normal or borderline LVEF and myocardial deformation results, as well as normal functional class and positive contractile reserve. None of the individuals analyzed exhibited late gadolinium enhancement or extracellular volume and T1 mapping alterations.
Interestingly, we found high penetrance of LAD, always with normal atrial contractility. This finding had not previously been associated with HCN4 variants. The mechanism underlying the LAD development is unknown. As previously explained, LAD could be a result of the anatomical structure, because HCN4 contributes to the embryonic development of the left ventricle and both atria,26 or a physiological adaptation to SB.
HCN4 variants with SSS, LAD, and LVNC implicationsThe association between HCN4 variants and the combined SSS, LAD, and LVNC phenotype seems undeniable. The noncompaction pattern exhibited by the patients with the present HCN4 p.R378C variant in this family suggests low aggressiveness and good prognosis. Left atrial remodeling is an important underlying substrate for AF and could be related to the appearance of the familial AF previously linked to HCN4 variants.17
The assessment and follow-up of patients with emerging HCN4 variants are not currently established. The follow-up that we have proposed for this family is mainly focused on the detection of systolic ventricular dysfunction and arrhythmic complications, mainly in phenotypes with LAD and the probability of AF development. We have decided to perform an annual clinical review with electrocardiography and Holter electrocardiography. The echocardiogram will be repeated every 1 to 2 years (except for changes in the clinical situation).
Study limitationsThis study included only 1 family and assessment of the entire family was not possible. However, a large number of family members has been evaluated. Indeed, our study reports the largest family linked to the HCN4 gene described to date and a comprehensive clinical study, which included cardiopulmonary exercise testing and CMR.
Our study, similar to others, does not explain the mechanism underlying the effect of the HCN4 variant on the development of left ventricular hypertrabeculation or LAD. Nonetheless, this is the first work involving the analysis of patients by CMR with parametric techniques and feature-tracking myocardial deformation, and we have shown the existence of a possible subclinical systolic dysfunction and LAD with potential clinical implications.
CONCLUSIONSThe combined phenotype of SSS, LAD, and LVNC is associated with different heritable HCN4 variants. Patients with familial forms of SSS should be studied to rule out the presence of structural heart disease, and HCN4 variants should be included in the genetic diagnosis, even individuals with SB and isolated LAD. In addition, HCN4 variants should be added to other recognized genes in the study of patients with LVNC. We report a family with a HCN4 c.1123C>T;(p.R375C) variant causing the combined phenotype of SSS, LAD, and LVNC with a benign course. Further studies are necessary to elucidate the pathophysiological mechanism of the atrial dilation and hypertrabeculation/noncompaction phenotype, as well as characterize the natural history of patients affected by HCN4 variants and the risk stratification in this specific population.
- -
Due to recent evidence, the association between HCN4 variants and the combined SSS, LAD, and LVNC phenotype seems undeniable, with phenotypes appearing to differ according to the underlying variant.
- -
The main complications related to HCN4 variants are arrhythmic complications, such as AF and sudden death, early pacemaker need, cardioembolic complications, and systolic ventricular dysfunction.
- -
We found high penetrance of LAD, always with normal atrial contractility, a novel finding for HCN4 variants. The importance of this result is that this structural abnormality could be related to the appearance of familial AF previously linked to HCN4 variants.
- -
The noncompaction pattern exhibited by patients with the present HCN4 p.R378C variant suggests low aggressiveness and good prognosis.
- -
We propose a follow-up of patients with HCN4 variants that is mainly focused on the detection of systolic ventricular dysfunction and arrhythmic complications, mainly in phenotypes with LAD and the probability of AF development.
The present study was financed in part by the Spanish Cardiovascular Center Network (CIBERCV), supported by Instituto de Salud Carlos III, Comunidad de Madrid (B2017/BMD-3738), and Ministerio de Economía y Competitividad (SAF2017-88116-P) to R. Caballero and E. Delpón. M. Alonso-Fernández-Gatta is funded by a Río Hortega contract (CM19/00055) supported by Instituto de Salud Carlos III (co-funded by the European Social Fund “Investing in your future”).
Conflicts of interestThe authors have no conflicts to disclose.
Supplementary data associated with this article can be found in the online version available at https://doi.org/10.1016/j.rec.2020.06.019