Keywords
INTRODUCTION
Sustained elevation of blood pressure (BP) is associated with a significant increase in cardiovascular morbidity and mortality in hypertensive patients. This is because arterial hypertension (AHT) can damage the structure and alter the function of the arteries, heart, brain, and kidney. Specifically, patients with AHT are at threat of developing a series of structural and functional alterations of the heart that constitute so-called hypertensive heart disease (HHD). Left ventricular hypertrophy (LVH) forms the macroscopic injury characteristic of HHD, but a series of microscopic changes underlie this giving rise to an entity known as myocardial remodeling. Cardiomyocyte hypertrophy and apoptosis, myocardial fibrosis, and intramyocardial artery and arteriole wall hypertrophy are the definitive structural elements of myocardial remodeling present in HHD.
MOLECULAR AND CELLULAR BASES
Like other organs, the heart consists of highly differentiated parenchymal cells, cardiomyocytes, and stroma formed by the extracellular matrix, tissue fluid, and undifferentiated multipotent mesenchymal cells. The cardiac extracellular matrix is mainly made up of fibrillar and non-fibrillar collagen, laminin and elastin fibers, proteoglycans and integrins. Fibrillar type I and III collagen molecules are the most abundant in adult heart and exhibit their typical triple helical shape due to the spatial orientation of their 3 α-polypeptide chains. Fibrillar collagen serves as a structural framework for cardiomyocytes and the intramyocardial vasculature, while conferring myocardial tissue with the stiffness that makes it resistant to distortion during the cardiac cycle. Furthermore, fibrillar collagen connects the contractile elements of adjacent cardiomyocytes, as well as acting as a transducer of cardiac muscle contraction toward the ventricular chamber. Although there is a shortage of fibrillar collagen in specific heart pathologies, most chronic heart diseases are characterized by an excessive focal (scar) or diffuse (fibrosis) accumulation of this.1
Studies carried out on post-mortem human heart2 and endomyocardial biopsies3 show that the myocardial collagen volume fraction (CVF, a measure of the quantity of collagen fibers accumulated in the myocardium) is significantly greater in HDD patients than in normotensive control subjects. Histologically, hypertensive myocardial fibrosis presents the following defining characteristics4-6: there is an initial excessive deposition of type III collagen fibers, followed by type I as the process progresses; the fibers are arranged as bundles lining the interstices and around the intramyocardial vessels; fiber accumulation is not limited to the left ventricle and is also present in the other cardiac chambers; the amount of fiber accumulation is inversely related to the number of cardiomyocytes and directly related to their degree of hypertrophy.
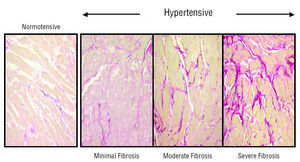
There are few studies on the prevalence of myocardial fibrosis in HHD. A study which established degrees of fibrosis based on comparing CVF values between normotensive subjects without LVH and hypertensive patients with LVH, verified that 11% of the patients presented null-minimum fibrosis, 58% mild-moderate fibrosis, and 31% severe fibrosis (Figure 1).3 Thus, fibrosis is a practically constant lesion in the myocardium of HDD patients.

Figure 1. Collagen content in the hearts of a normotensive patient and 3 patients with hypertensive heart disease. Patients are classified according to the degree of myocardial fibrosis: minimum (left), moderate (center), and severe (right). Sections are stained with picrosirius red and collagen fibers appear red.
CAUSAL MECHANISMS
The excess of myocardial collagen fibers in HHD is the result of a combination of an increase in collagen synthesis, by fibroblasts and myofibroblasts, and a decrease or absence of change in its degradation by matrix metalloproteinases.7 This hypothesis is supported by experimental findings that show overexpression of procollagen type I genes8 (collagen type I precursor)8 and reduced collagenase (enzyme governing collagen type I degradation)9 activity in hypertrophied left ventricle of spontaneous hypertensive rats (SHR). The combination of different factors (hemodynamic, hormonal, genetic and environmental) can give rise to this imbalance.
Hemodynamic Factors
In vivo experiments have shown that chronic pressure overload stimulates both gene expression and collagen protein synthesis in the myocardium, which favors excess deposition of collagen fibers and the resulting fibrosis.10 Furthermore, in vitro studies have shown that procollagen type I synthesis is stimulated in cardiac fibroblasts under mechanical cyclic overload,10 like that produced under AHT conditions. Thus, hemodynamic left ventricular overload due to AHT can favor myocardial fibrosis.
Several clinical observations support this possibility. Tanaka et al11 found that CVF increased from the exterior to the interior of the left ventricular free wall in hypertensive human hearts, which is probably a reflection of the transmural gradient of parietal stress. Rossi2 reported that when hypertensive patients' hearts were grouped according to their weight, CVF increased progressively by weight. Furthermore, there was an association between the severity of interstitial fibrosis and that of LVH. Finally, our group reported that systolic BP and pulse pressure were higher in the patients with severe fibrosis than in the patients with minimum and moderate fibrosis.3
Non-Hemodynamic Factors
Two types of findings indicate that, in addition to hemodynamic factors, non-hemodynamic factors can also contribute to the development of myocardial fibrosis in AHT. As mentioned, the first refers to the presence of myocardial fibrosis not only in the left ventricle but also in the right,4,12 in interventricular septum13 and left atrium,14 as reported in post-mortem studies of hearts from HDD patients. Second, recent studies have shown that the ability of antihypertensive treatment to regress myocardial fibrosis in hypertensive patients is independent of its antihypertensive efficacy.15,16 Thus, the current view is that the development of myocardial fibrosis can also be a consequence of the hormonal factors that stimulate fibrillar collagen metabolism predominating over those which inhibit it (Table 1).7 Among these, the renin-angiotensin-aldosterone system (RAAS) agonists are especially relevant.

Thus, clinical studies where angiotensin-converting enzymes (ACE) are inhibited15,17 or angiotensin II (ANG II) type I receptors (r-AT1) are blocked16,18 have indicated the relevance of ANG II in the development of myocardial fibrosis in HDD patients. In fact, many experimental studies show that the interaction of this peptide with r-AT1 has multiple profibrotic effects in the heart, including inducing fibroblast hyperplasia and cardiomyocyte and myofibroblast differentiation, fibrillar collagen synthesis activation and collagen fiber degradation inhibition.19 In addition, various findings indicate that interactions between factors produced by cardiomyocytes (e.g., osteopontin), macrophages (e.g., plasminogen activator inhibitor 1), and fibroblasts (e.g., transforming growth factor-beta) would mediate the profibrotic effects of ANG II.20 Furthermore, fibrosis could be part of a reparative response to the inflammation and oxidative stress induced by ANG II through interacting with r-AT1 located in cardial microvasculature cells.21
Aldosterone is another hormonal factor that can be relevant in the development of myocardial fibrosis. Chronic aldosterone infusion in uninephrectomized rats fed with a high-salt diet is associated with marked accumulation of collagen fibers in both heart ventricles.22 In this model, cardiac fibrosis is prevented by spironolactone, a mineralocorticoid receptor blocker,23 and so the fibrotic mechanism of aldosterone would involve interacting with this receptor present in cardiac fibroblasts and myofibroblasts.24 In addition, activation of the mineralocorticoid receptor can facilitate the profibrotic action of ANG II via upregulation of r-AT1 expression.25 It is relevant to note that the profibrotic action of aldosterone seems to be independent of BP, given that mineralocorticoid receptor inhibition with eplerenone reduces the myocardial fibrosis produced in mice with chronic pressure overload in the absence of significant changes in systemic BP.26
Genetic and Environmental Factors
Some findings indicate that genetic factors play a role in modulating hypertensive myocardial fibrosis. A microsatellite marker for rat ACE gene has been identified, making it possible to differentiate the alleles of this gene and its association with different degrees of enzyme activity in plasma.27 Higher degrees of ACE activity in the left ventricle and a more extensive development of ventricular fibrosis in response to isoproterenol have been found in rats carrying allele B than in rats carrying allele L treated with the same compound.28 On the other hand, in a recent study,29 our group analyzed the influence of A1166C polymorphism of the r-AT1 gene on the ability of losartan to inhibit collagen type I synthesis and regress myocardial fibrosis in HDD patients. The patients were genotyped for this polymorphism and divided into 2 subgroups: AA and AC/CC. Collagen synthesis was significantly greater in the AA patients and decreased with losartan treatment more than in the AC/CC patients. Although the molecular bases for this association are not very clear, they may be related to changes in RAAS cardiac activity.
In the same way that it is known that excess salt intake facilitates the development of LVH in animals and humans with AHT, regardless of its effects on BP, recent experimental findings indicate that this could also be applicable to myocardial fibrosis. Thus, a recent study found that increased salt intake was associated with the development of biventricular myocardial fibrosis in SHR rats, but not in normotensive Wistar-Kyoto rats; furthermore, there was no association between elevated BP and increased CVF in SHR.30 These findings indicate the potential for myocardial fibrosis of the hypertensive genetic substrate interacting with the exogenous factors commonly linked to AHT.
CLINICAL CONSEQUENCES
As shown in Figure 2, myocardial fibrosis can contribute to ventricular dysfunction, reduced coronary flow reserve and ventricular arrhythmias adversely affecting cardiovascular outcomes in HDD patients.31

Figure 2. Mechanisms by which myocardial fibrosis contributes to the complications of hypertensive heart disease.
Ventricular Dysfunction
The content of myocardial collagen confers resistance to stiffness during diastole.32 Furthermore, myocardial elasticity during contraction depends inversely on collagen content.32 Various clinical and experimental trials have shown that fibrosis increases stiffness and reduces the elasticity of myocardial tissue.33 Specifically, a two- or threefold increase in CVF adversely affects diastolic stiffness (promoting diastolic dysfunction), whereas a fourfold increase in CVF or more is associated with an additional increase in diastolic stiffness and a reduction in systolic elasticity (promoting systolic dysfunction).
Several clinical findings support this. Recently, our group has shown that there is a direct association between myocardial collagen content and the stiffness of the left ventricular chamber in HHD patients (Figure 3A), and that regression of the severe fibrosis induced by losartan in these patients is accompanied by reduced myocardial stiffness.18 Sugihara et al34 found that CVF was the most relevant factor associated with diastolic dysfunction in hypertensive patients. Brilla et al15 found that the reduction in CVF after chronic treatment with the ACE inhibitor lisinopril was associated with improved left ventricular diastolic function in AHT patients. Our group35 (Figure 3B) and McLenachan and Dargie36 have reported an inverse association between CVF and ejection fraction (EF) in HDD patients. Finally, studies done in patients with HDD-associated heart failure have found an association between reduced myocardial fibrosis and improved cardiac function in such patients.37

Figure 3. A: direct correlation of collagen fraction volume (CVF) with left ventricular chamber stiffness (KLV) in patients with hypertensive heart disease. B: inverse correlation of CVF with ejection fraction (EF) in patients with hypertensive heart disease. Panel A taken from Díez et al.18 Panel B taken from Querejeta et al.35
Reduced Coronary Flow Reserve
HHD patients can present symptoms and signs of myocardial ischemia, although the coronary arteries may appear normal under angiography.38 Reduced coronary flow reserve is probably the cause of myocardial ischemia in these cases.39
Functional and structural alterations in the coronary microcirculation have been reported in HHD that can be associated with reduced coronary flow reserve, including endothelial dysfunction, media layer thickening with reduced lumen and collagen fiber accumulation in the periarteriolar area.40 It has been reported that chronic treatment with the ACE inhibitor perindopril induces an increase in coronary flow reserve in hypertensive patients associated with significant periarteriolar fibrosis regression and a slight, although nonsignificant, reduction in hypertrophy of the arteriolar media layer.18
Isoyama et al41 have shown the relevance of periarteriolar collagen in impaired coronary flow reserve in experimental studies. It was found that, after declamping the aorta, normalized BP induced regression in the hypertrophied arteriolar media layer; however, coronary flow reserve was only normalized after collagen accumulation in the adventitia was inhibited with β-aminopropinitril. Thus, it can be assumed that perivascular fibrosis is a limiting factor regarding intramyocardial vessel distensibility in HDD patients.
Ventricular Arrhythmias
Epidemiological studies, such as the Framingham study,42 have shown a high incidence of ventricular arrhythmias in HDD patients. Arrhythmias are associated with greater mortality and sudden death in these patients.
McLenachan and Dargie36 analyzed the possible correlates of ventricular arrhythmias in HHD patients and found that patients with arrhythmias had greater left ventricular mass and CVF values than patients without arrhythmias. Ejection fraction and the number of coronary vessels with significant stenosis (>50%) were similar in the 2 groups of patients. Thus, the high incidence of arrhythmias in HHD patients cannot be attributed exclusively to coexistent coronary artery disease or left ventricular dysfunction, but can be related to fibrosis and the adaptive phenotypical changes in LVH-associated cardiomyocytes.
Fibrosis could cause arrhythmias via anatomical uncoupling due to myocardial heterogeneity and via a reentry mechanism generated by the zigzag propagation of the transverse waveform.43
DIAGNOSTIC APPROACH
Due to the adverse effects on the heart that myocardial fibrosis can have in HHD patients, its assessment can help in diagnosing myocardial remodeling and establishing its prognosis in these patients. Furthermore, being able to assess fibrosis can be useful when evaluating the effects of antihypertensive treatment on remodeling.
Histological Diagnosis
Given that myocardial fibrosis is a histopathological lesion, the most reliable diagnostic method would be endomyocardial biopsy. In general, the endomyocardial biopsy procedure is not technically complex and is clinically safe for the patient.44 This assertion is especially relevant taking into account that fibrosis existing in the interventricular septum has been shown to be representative of that on the left ventricular free wall,13 which means that the risk of complications is further reduced by biopsy of the septum via the right ventricle using a venous approach. Nevertheless, it should be recognized that endomyocardial biopsy is an invasive technique that, due to its technical requirements, presents obvious limitations regarding its widespread application.
Diagnosis Via Imaging Methods
Ultrasonic characterization of myocardial tissue enables the identification and characterization of its physical condition by analyzing the interactions between ultrasounds and the tissue itself. This technique is based on the principle whereby the interaction of the ultrasound waves with normal tissue causes ultrasonic signal reflections that have some specific characteristics that can be quantified (e.g., via tissue-integrated backscatter), which makes it possible to define the myocardial structure and functional qualities. These characteristics differ from those observed when ultrasounds interact with abnormal tissue, such as fibrotic tissue.45 In this context, an association between alterations in echoreflectivity has recently been shown as a reduction in cyclic variation in the backscatter signal and the increase in CVF in the hearts of HDD patients.46,47 However, reproducing the information provided by these methods may encounter difficulty. Furthermore, due to its high cost, its use in normal clinical practice would be restricted to centers where these methods are available.
Magnetic resonance imaging (MRI) is another promising technique for characterizing myocardial composition, especially when obtained with gadolinium, which has been validated and seems useful for the quantification of non-ischemic fibrosis. A study of patients with right ventricular arrhythmogenic myocardiopathy found a correlation between the parameters measured using this technique and histologically determined myocardial fibrosis.48 Magnetic resonance imaging has proved useful in detecting the fibrosis present in endomyocardial disease.49
Biochemical Diagnosis
In recent years, alternative methods to previous ones have been developed based on the immunochemical determination of peptides derived from the metabolism of collagen type I and III present in the blood (Table 2). Of all the peptides studied, only one, the carboxy-terminal propeptide of procollagen type I (PICP), meets the requirements to consider it both a circulating marker of cardiac collagen type I synthesis and a myocardial fibrosis biomarker.50

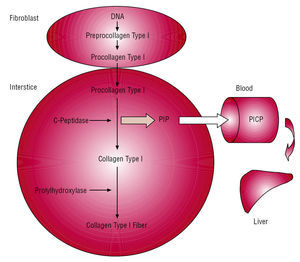
The method is based on the following (Figure 4): cardiac fibroblasts and myofibroblasts secrete the procollagen type I precursor molecule into the interstitial space. This is converted into the final collagen type I fiber-forming molecule due the action of specific proteinases that hydrolyze the terminal peptides of the precursor. Specifically, a specific carboxypeptidase hydrolyzes PICP that, via the cardiac venous and lymphatic systems, flows into the systemic circulation. One molecule of PICP appears in the blood for each procollagen type I molecule that is transformed into one collagen type I molecule, where it can be detected via a specific radio-immunoassay (RIA) or enzyme-linked immunosorbent assay (ELISA).

Figure 4. Formation and passage into blood of propeptide carboxy-terminal of procollagen type I (PICP) released during conversion of procollagen type I to collagen type I. Taken from López et al.50 PICP indicates propeptide carboxy-terminal of procollagen type I.
Pilot studies conducted by our group have shown that PICP serum concentrations are abnormally higher in HDD patients3 and SHR,51 and that in both cases serum concentrations of this peptide are directly correlated with CVF (Figure 5A). We have recently shown that PICP concentrations increase progressively as HDD evolves, reaching their highest concentrations in heart failure (CHF) patients, and have a direct correlation with the myocardial content of collagen type I.35 Furthermore, in the same study, we proved the cardiac origin of PICP, given that in hypertensive patients there is a gradient between PICP values measured in coronary blood and those in peripheral blood (Figure 5B), as well as a direct and highly significant correlation between the two. Finally, in other studies we have reported that PICP values and the amount of myocardial fibrosis are modified in parallel under antihypertensive treatment both in SHR7 and in patients with CHF and normal heart function16,18 and in patients with CHF and HDD.37 Although preliminary, these data indicate that the PICP present in the peripheral blood of HHD patients is essentially of cardiac origin and provides a reliable index of the amount of collagen type I fibers present in the myocardium, as well as changes in the amount of fibrosis induced by the treatment.

Figure 5. A: direct correlation of collagen volume fraction (CVF) with propeptide carboxy-terminal of procollagen type I (PICP) serum concentrations measured in peripheral blood. B: direct correlation of PICP serum concentrations measured in coronary sinus and peripheral blood. Panel A taken from Querejeta et al.3 Panel B taken from Querejeta et al.35
In the same way that cerebral and atrial natriuretic peptides are considered biomarkers of systolic dysfunction in CHF patients, it would be interesting to explore whether PICP can be of value as a biomarker of structural myocardial damage in these patients. In fact, we could infer this from the observation that serum concentrations of PICP are significantly higher in HHD and CHF patients with depressed EF than in patients with preserved EF.52 However, such differentiation would not be valid for patients with heart failure deriving from other etiologies, above all those with ischemic heart disease.53
THERAPEUTIC ASPECTS
The time may have come to propose that the treatment of hypertensive patients should not focus exclusively on normalizing BP, but should also have as an objective the prevention or correction of structural and functional alterations in AHT target organs. With regard to HHD, the European Society of Hypertension and the European Society of Cardiology state in their guidelines on treating hypertension that "Future studies should investigate treatment-induced effects on indices of collagen content or fibrosis of the ventricular wall, rather than on its mass only."54 From this perspective, drugs with the ability to repair myocardial fibrosis will be, basically, those which reestablish equilibrium between the factors that stimulate and those that inhibit the metabolism of collagen type I and type III molecules.55
Antihypertensive Drug Findings
This cardioreparative concept has been proven clinically in several prospective studies of limited size where biopsies were used to quantify myocardial fibrosis. Brilla et al15 showed that treating HHD patients with lisinopril reduces myocardial fibrosis independently of BP monitoring and LVH regression, whereas treatment with hydrochlorothiazide does not have such an effect. Fibrosis reduction is associated with improved left ventricular diastolic function. Our group has shown that 1 year of losartan treatment is associated with reduced PICP serum concentrations and reduced CVF in HDD patients.18 However, patients treated with amlodipine do not show significant changes in either parameter, despite having similar antihypertensive efficacy.16 In a later study, we found that the ability of losartan to induce regression of severe fibrosis in HHD patients is independent of its ability to reduce BP or left ventricular mass and is associated with reduced ventricular chamber stiffness.18 Taken together, these data confirm what is found in SHR, where it has been verified that the pharmacological interference on the action and production of ANG II is effective in the regression of cardiac fibrosis, independent of its antihypertensive effect.8,56
Recently, we reported that HDD/CHF patients undergoing chronic treatment with torasemide present greater reductions in serum PICP concentrations and CVF and greater improvement in functional class than patients treated with furosemide (Figure 6).37 Unlike furosemide, it is noteworthy that torasemide is able to inhibit the adrenal secretion of aldosterone57 and its bonding to mineralocorticoid receptor,58 as well as decreasing transcardiac extraction of aldosterone in CHF patients.59 Based on this, it could be hypothesized that iton cardiac fibrosis occur by inhibiting the profibrotic actions of aldosterone.

Figure 6. Effects of chronic treatment with torasemide or furosemide on collagen volume fraction (CVF), propeptide carboxy-terminal of procollagen type I (PICP) serum concentrations, and functional class (according to New York Heart Association classification) in heart failure patients. Data are presented as mean ± standard error of the mean. The dark and light columns represent the values before and after treatment, respectively. Adapted from López et al.37
Findings With Other Compounds
Several experimental works have explored alternative therapeutic strategies to reduce myocardial fibrosis. Thus, it has been shown that tranilast [N(3,4-dimethoxycinnamoyl) anthranilic acid]60 and Ac-SDKP (N-acetyl-seryl-aspartyl-lysyl-proline)61 reduce inflammation and cardiac fibrosis in rats with experimental hypertension via mechanisms probably related to TGF-β inhibition. It has also been found that fenofibrates-peroxisome proliferators-activated receptor-α (PPAR) activators-reduce myocardial fibrosis in mineralocorticoid-induced hypertensive rats, probably by preventing inflammatory mediator release associated with the NF-κB pathway.62 Finally, the proteasome inhibitor MG132 has been shown to eliminate expression of collagen molecules in isolated fibroblasts and reduce myocardial fibrosis in SHR.63 Although this group of findings opens new perspectives on the treatment of myocardial fibrosis, additional studies are needed before implementing their therapeutic use in HHD.
Findings Via Other Procedures
Recently, it has been reported that cardiac resynchronization therapy reduces CVF in patients with CHF of varying etiology.64 A recently published study reported that the beneficial effects of cardiac resynchronization therapy on cardiac morphology, function and performance are associated with reduced PICP serum concentrations in CHF patients.65 The specific mechanisms by which resynchronization reduces collagen type I synthesis and deposition in the myocardium of these patients remains to be clarified.
On the other hand, experimental studies have shown that intracardiac injection of human mesenchymal stem cells reduces fibrosis in animals with myocardial infarction,66 probably by releasing factors that inhibit the metabolism of fibrilar collagen.67
CONCLUSIONS
Myocardial fibrosis forms the histomorphological substrate of HDD. Available data indicate that the RAAS is determinant in myocardial fibrosis developing in HHD. Fibrosis can contribute to the transition of LVH to congestive heart failure in hypertensive patients, as well as to developing other complications specific to HHD. From this point of view, the clinical treatment of these patients should involve something more than the diagnosis and normalization of AHT and LVH. A more thorough approach would include measures also aimed at detecting and treating myocardial fibrosis. Some preliminary evidence indicates that measuring serum PICP can prove useful in diagnosing myocardial fibrosis in HDD patients. On the other hand, evidence already exists showing that the aim of reducing myocardial fibrosis is achievable in HDD patients, mainly via the use of drugs that interfere with the RAAS. Taken together, this information forms the basis for undertaking large, long-term clinical trials to clarify whether the diagnosis and regression of myocardial fibrosis helps to improve the prognosis and evolution of HHD patients.
Correspondence: Dr. J. Díez Martínez.
Área de Ciencias Cardiovasculares. Edificio CIMA. Facultad de Medicina.
Avda. Pío XII, 55.
31008 Pamplona. Navarra. España.
E-mail: jadimar@unav.es