Keywords
INTRODUCTION
Left ventricular cardiomyopathy and ischemicheart disease have typically been considered theprimary causes of ventricular arrhythmias (VAs)and sudden cardiac death (SCD). However,arrhythmias originating in the right ventricle(RV) have attracted the attention of scientists over the last two decades for several reasons.VAs originating in the RV usually affect youngerpatients and can potentially lead to SCD. Thepathophysiologic mechanism underlying thesearrhythmias has not been completely elucidated,leaving room for active research and differinginterpretations. Genetics are also increasinglyhelping to explain the pathogenetic, diagnosticand prognostic aspects of some of thesearrhythmias.
ARRHYTHMOGENIC RIGHT VENTRICULARCARDIOMYOPATHY/DYSPLASIA
Arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVD) is characterized by VAs andprogressive structural abnormalities of the RV.1,2 Myocardial degeneration may extend to the leftventricle, especially in advanced stages of the disease.3 ARVD can exist in both sporadic and familial forms.The disease is characterized by either massive orpartial progressive replacement of myocardium byfatty or fibro-fatty tissue. This infiltration providesa substrate for electrical instability and leads toVAs ranging from isolated premature ventricularcontractions (PVC) to sustained VTs or ventricularfibrillation.3,4 The prevalence of the disease in thegeneral population has been estimated to range from1 in 2000 to 1 in 10 000. Eighty percent of cases arediagnosed in patients under 40 years of age. ARVDshould be suspected in all young patients with anapparently normal heart presenting with syncope,VT, or cardiac arrest. As will be discussed, the diseaseshould also be suspected in athletes presenting withsymptoms indicative of arrhythmias (palpitations,syncope).
Genetic Aspects
ARVD is a heritable condition.5 There is ahigh familial incidence (30%-50% of cases) withautosomal-dominant inheritance, various degreesof penetrance, and polymorphic phenotypicexpression. An autosomal-recessive form ofARVD has also been described which is associatedwith palmoplantar keratoderma and woolly hair(Naxos disease).6 This type of ARVD is caused bya mutation in the plakoglobin gene, the product ofwhich is a component of desmosomes and adherensjunctions.7 The first mutation causing nonsyndromalARVD was reported by Rampazzo et al in 2002.8 The mutation was found in the desmoplakin gene,encoding a component of the desmosome. In 2004,Gerull et al described 25 mutations in the cardiacdesmosomal gene plakophilin 2.9 Desmocollin 2and desmoglein 2, other putative disease-causingmutations in plakoglobulin and desmoplakin, as wellas in other desmosomal genes, were later identifiedin patients with nonsyndromal ARVD.10 Nowadays,desmosomal dysfunction is considered the finalcommon pathway of ARVD pathogenesis.
Different genetic variants of ARVD havebeen mapped and over 140 disease-causingARVD mutations have been published, the vastmajority encoding desmosomal proteins. Somenondesmosomal genes have also been associatedwith autosomal dominant ARVD, such as thetransforming growth factor b-3 gene (TGF b3),11 which modulates the expression of cell-contactproteins and the ryanodine receptor 2 (RyR2)gene.12 First described in 8 families, the RyR2encodes receptors involved in calcium release fromthe sarcoplasmic reticulum. However, opinions stilldiffer as to whether patients with RyR2 mutationsshould be considered as suffering from ARVD orfrom a cathecolaminergic polymorphic VT. Themost recently described mutation in the TMEM43gene is responsible for a highly lethal and fullypenetrant ARVD variant (ARVD5).13
Desmosomes and ARVD
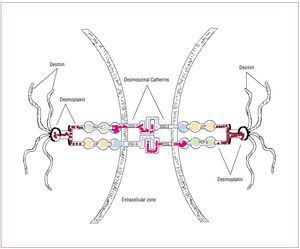
The structural and functional integrity ofcardiac tissue is supported by desmosomes,adherens junctions and gap junctions located atintercalated disks (Figure 1). Desmosome integrityis required to maintain the normal function of gapjunctions as intercellular channels responsible forthe electrical coupling and signaling mechanismsin the regulation of cell growth, differentiationand development.

Figure 1. In desmosomes, junctional plakoglobin (JUP), plakophilin (PKP)-2, and desmoplakin (DSP) anchor desmin intermediate filaments to desmosomalcadherins, securing the mechanical cell-cell adhesion. Desmosomal cadherins, desmoglein (DSG)-2 and desmocollin (DSC)-2 are transmembrane proteinsforming an extracellular zipper-like dimer with the corresponding part of desmosomal cadherins of the adjacent myocardial cell. Modified from Sen-Chowdhryet al.14
Genetic mutations responsible for ARVDresult in haploinsufficiency and reducedexpression of desmosomal proteins, which may predispose mechanical cell contacts to rupture,potentially triggered by mechanical stress of theRV (such as that occurring during exercise orsports activity). Degeneration and death of thecardiomyocytes is the pathological consequenceof these mutations in adhesion proteins, with thesubsequent progressive replacement by fatty andfibro-fatty tissue.
Structural Abnormalities
ARVD is characterized by replacement ofmyocardium with fatty and fibrous tissue thatinitially affects the epicardium and then theendocardium. This process most commonlyinvolves the posterior and inferior areas of the RV inflow tract adjacent to the tricuspid valve,but it also affects the anterior infundibulum andthe apex, thus forming what is known as the "triangle of dysplasia."5 Loss of myocardium mayresult in focal thinning of the ventricular free wall,focal bulging of the RV wall in diastole, and rightventricular outflow tract (RVOT) enlargement.Functionally, the disease may result in global orregional contraction abnormalities, RV systolic/diastolic function, RV aneurysm formation, and RVdilatation and hypokinesis.15 The interventricularseptum is generally spared. Endomyocardialbiopses, which are usually obtained from theseptum, may therefore be non-diagnostic.Endomyocardial biopsy performed in the RV haslow sensitivity because of the segmental nature of fatty infiltration. Samples should be retrieved fromthe RV free wall, as the fibro-fatty replacement isusually transmural and thus detectable from theendocardial approach. The risk of perforationduring RV endocardial biopsy in the apex and freewall of the RV should not be underestimated.
ARVD should be distinguished from Uhl's disease,a rare congenital disorder in which right ventricularmyocardium is absent, resulting in a paper-thin rightventricular wall.15
Clinical Presentation
ARVD usually presents with VTs with left bundlebranch block (LBBB) morphology, originating fromthe RV, in apparently healthy adolescents or youngadults. The VAs may be asymptomatic and detectedby routine electrocardiogram, or they may causepalpitations, syncope or SCD. It has been estimatedthat ARVD accounts for up 5%-20% of SCD inindividuals under 35 years of age.15 However, theclinical manifestations of the disease may varywidely in time of onset and degree of severity. Theinteraction between different genetic substrateswith various degrees of penetrance and the presenceof external triggers (such as strenuous exercise orvigorous training) explains the broad spectrum ofclinical presentations, which can include palpitations, fatigue, atypical chest pain, syncope, and SCD. Ageat the time of the first manifestation ranges between15 and 35 years. Men are more frequently affectedthan women and usually present with more extensivedisease. Symptomatic heart failure is an unusualmanifestation of ARVD and usually occurs in moreadvanced stages of the disease. Patients with a longhistory of ARVD frequently show involvement of theleft ventricle with clinical symptoms of biventricularheart failure.16
Diagnosis
According to McKenna et al's 1994 ARVDTask Force Report, diagnosis of ARVD should bebased on the presence of structural, histological,electrocardiographic, arrhythmic, and geneticfactors as well as on family history (Table 1). Patientsmust have either 2 major criteria, 1 major and 2minor criteria, or 4 minor criteria to be consideredas affected by ARVD.17 A modification of the TaskForce Criteria for the diagnosis of ARVD wasproposed in 2002 to take into account the case of firstdegree family members of a proband. In the lattercase, the presence of right precordial T wave inversion(V2-V3), late potentials on signal-averaged ECG,or VT with LBBB morphology, or mild functionalor morphological changes of the RV on imaging, should be considered major criteria and diagnosticfor familial ARVD.18 A further modification ofthe diagnostic criteria19 was published recently,in an attempt to increase diagnostic sensitivity bytaking into account emerging diagnostic modalitiesand advances in the genetics of ARVD, and byfurnishing quantitative parameters (Table 2). Thus,routine assessment should include 2-dimensionalechocardiography, electrocardiography, signal-averaged electrocardiography, 24-hour Holtermonitoring, stress test, and family history. Whenthe results are inconclusive, additional imagingexaminations and endomyocardial biopsy should beconsidered.


Electrocardiographic Abnormalities
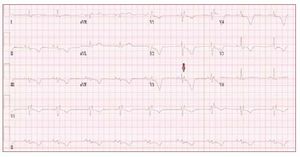
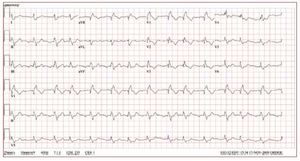
The electrocardiogram in patients with ARVDusually shows a regular sinus rhythm, with a QRSduration over 110 ms in lead V1. Electrocardiographicchanges include inverted T waves in the rightprecordial lead beyond V1 in the absence of rightbundle branch block (RBBB) and right ventricularlate potentials in the form of "epsilon waves" inleads V1-V3 (Figure 2).

Figure 2. The ECG is from a 21-year-oldmale with arrhythmogenic right ventriculardysplasia. Right bundle branch block,epsilon wave (arrow) and diffuse T waveinversion are present.
T wave inversion in leads V1-V3 is a well recognizedECG feature of ARVD and in the absence ofRBBB is currently proposed as a major diagnosticcriterion.19 The juvenile pattern of T wave inversionin V1-V3 or beyond is a normal variant in childrenunder 12 years of age. This variant is present in 1%-3% of the healthy population aged 19 to 45 yearsand 87% of patients with ARVD. Epsilon wavesare "postexcitation" electrical potentials of smallamplitude that occur in the ST segment after theend of the QRS complex. Epsilon waves, which areseen in 33% of patients with ARVD, are considereda major diagnostic criterion for ARVD. Different ECG abnormalities may be present according todisease extension. However, the presence of a normalECG should not preclude diagnosis of a preliminaryform of ARVD.20
Imaging Evaluation
Imaging techniques for diagnosingmorphofunctional abnormalities consistentwith ARVD include conventional angiography,echocardiography, computed tomography,radionuclide angiography, and magnetic resonanceimaging (MRI). Right ventricular angiography hashistorically been regarded as the best imaging testfor the diagnosis of ARVD and has been shownto be highly specific (90%).21 Echocardiography isless invasive and represents the first-line approachin evaluating patients with suspected ARVD orin screening family members. MRI has the abilityto differentiate fat from muscle and allows for ahighly accurate and quantitative evaluation of RVsize and function. Although the utility of MRI inARVD diagnosis is well recognized, this techniquesometimes leads to over-diagnosis of ARVD. Thesensitivity and specificity of MRI for detecting RVintramyocardial fat in the diagnosis of ARVD isvariable, ranging from 22% to 100%.22-25 Identifyingfat can be challenging because the RV is a thinstructure and areas of affected myocardium can bequite small. Moreover, it is now well known thatit may be normal for fat to be present in the RVmyocardium. Distinguishing pathologic adiposeinfiltration in areas where adjacent epicardial fat isnormally present, such as in the AV groove and theantero-apical portion of RV, may be particularlydifficult. Isolated areas of fat replacement have alsobeen reported in elderly patients, in those with long-term steroid use, in obese individuals, in those with other cardiomyopathies, and in idiopathic RVOTVT.26 Physicians should therefore avoid a diagnosisof ARVD when the only structural abnormalitiesare those seen on MRI.
Late-gadolinum-enhanced MRI can localize fibro-fatty changes in the RV myocardium. The fibro-fattyvariety of ARVD is more common than fatty ARVD.Thus, a potential role of late-gadolinum-enhancedMRI may be to detect early and subtle cases ofARVD which may otherwise be misdiagnosed.27,28 Documentation of fibrous tissue has been suggestedto be diagnostically more important than finding fatalone.29
Voltage Mapping: an Important DiagnosticTool
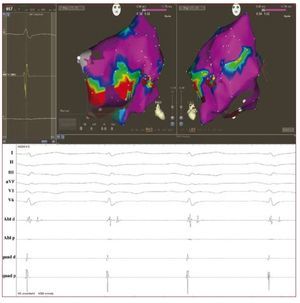
Emerging data suggest an increasing role forendocardial voltage mapping in identifying thepresence of scarring in the RV in early phases of thedisease. The technique has the potential to accuratelyidentify the presence, location and extent of thepathologic substrate of ARVD by demonstration of low voltage RV regions, ie, electroanatomicalscarring (Figure 3). In ARVD patients, RV scarringidentified with electroanatomical mapping hasbeen shown to correspond to areas of myocardialdepletion and to correlate with the histopathologicfinding of myocardial atrophy and fibro-fattyreplacement at endomyocardial biopsy.30 Corrado etal demonstrated that voltage mapping can be usefulin the differential diagnosis between RVOT-VT andthe early form of ARVD. In fact, in patients with VAsfrom the RV and an apparently normal RV usingconventional imaging, the presence of an area of lowvoltage regions in the RVOT identified a subgroup athigh risk for VT recurrence and SCD during clinicalfollow-up.31 Very recently, Wijnmaalen et al showedthat patients with scar-related right ventriculartachycardia with or without an established ARVDdiagnosis according to the Task Force criteria hada higher VT recurrence rate than patients withoutelectroanatomical scarring in the RV.32 These dataconfirm that voltage mapping might be an importantdiagnostic tool with prognostic and therapeuticimplications.

Figure 3. Above: voltage mappingreconstruction of the right ventricle insinus rhythm using an electroanatomicalmapping system (Carto 3, BiosenseWebster, USA) in a 43-year-old man withan overt form of arrhythmogenic rightventricular dysplasia and a symptomaticmonomorphic ventricular tachycardia.Areas of scar are identified (localvoltage <1.5 mv) at the right ventricleantero-lateral wall (basal part). Red dotsrepresent the radiofrequency ablationline delivered with the intent to block thescar-related macro reentry circuit. Below:endocavitarial late potentials recordedfrom the mapping catheter in and aroundthe scar regions
Differential Diagnosis
The main differential diagnoses of ARVDare RVOT-VT, sarcoidosis, idiopathic dilatedcardiomyopathy, and isolated myocarditis. BothRVOT-VT and ARVD occur in young, apparentlyhealthy individuals, and both may present with PVCsor VT with a LBBB and inferior axis. Althoughit is not difficult to diagnose a manifest case ofARVD, differentiation of ARVD at its early stagesfrom RVOT tachycardia, a usually benign andnonfamilial arrhythmic condition, remains a clinicalchallenge. If clinical doubts remain after traditionalexaminations (ECG, Holter) and imaging techniques(echocardiogram, right ventricular angiography,MRI), RV voltage mapping seems to be a usefulemerging tool to differentiate between the 2 entities.
Role of Genetic Analysis
A key clinical application of genetic analysisincludes confirmatory testing of proband cases tofacilitate interpretation of borderline investigationsand cascade screening of relatives.14,33 Genotyping relatives before a malignant clinical phenotypeis manifest may be crucial in preventing SCD.Management of relatives with non-overt forms ofARVD diagnosed by molecular genetic analysis mayprevent SCD in these subjects through serial clinicalfollow-up (electrocardiogram, echocardiogram,24-hour Holter, early recognition of symptoms),lifestyle modifications (restriction from extremeactivity), and prophylactic therapy when needed(antiarrhythmic drugs, implantable cardioverterdefibrillators [ICD]). Despite its reliability, geneticsequencing is an effort- and cost-intensive process,especially in the investigation of ARVD, in whichpotentially large numbers of genes are involved.
Very recently, a new diagnostic test basedon immunohistochemical desmosomal analysisof human myocardial samples obtained byendomyocardial biopsy has been reported in a smallpatient population.34 This study showed that theimmunoreactive signal level for the desmosomalprotein plakoglobulin was reduced at intercalateddisks in patients with ARVD. Interestingly,plakoglobin signal levels were reduced not onlyin right ventricular myocardium showing typicalpathological changes of fibro-fatty replacementbut also in normal-appearing left ventricle andinterventricular septum. Moreover, this was notobserved in others forms of heart-muscle disease.
Risk Stratification
The available data suggest that severe RV dysfunction,left ventricle involvement, syncope, young age, male sex, prior cardiac arrest, fast and poorly tolerated VTwith different morphologies, and familial occurrence ofjuvenile sudden death are the major factors in determininga poor outcome.35,36 High-risk patients present withclinical signs of right heart failure and/or have leftventricular dysfunction and a history of VT.36 These patients should be regarded as candidates for aggressivetherapeutic management. Conversely, patients withoutVT are at very low risk of cardiac events. Debate isstill on-going as to whether electrophysiological studyis useful37 or not38,39 in predicting the occurrence ofVT during follow-up. Lifestyle recommendations, i.e.restrictions on vigorous physical activity, may improvethe long-term outcome.
Treatment
The main goal of a management strategy is toprevent SCD. Currently, antiarrhythmic drugs,catheter ablation, and ICDs are the 3 main therapiesavailable for patients with ARVD.
Antiarrhythmic Drugs
Patients with ARVD and no history of syncopeor cardiac arrest, but with PVCs, couplets or shortventricular runs do not usually have an increasedrisk of arrhythmias and therefore do not requirespecific antiarrhythmic treatment. In patients withsustained VT, the aim of antiarrhythmic drugtherapy is not so much the suppression of VTrecurrences but the prevention of SCD. Sotalolat a dosage of 320-480 mg/day was identified asthe most effective drug, resulting in a 68% overallefficacy rate.40 Interestingly, a recent study in acohort of rigorously characterized ARVD patientsshowed that beta blockers were neither harmfulnor protective against clinically relevant VAs, thatsotalol was not effective, and that amiodarone hadthe greatest efficacy.41 Class I antiarrhythmic drugsproved effective in only a minority of patients withARVD. To date, prospective, randomized studieson antiarrhythmic drug efficacy in ARVD are notavailable.
Catheter Ablation
Current indications for catheter ablation inpatients with ARVD include monomorphic and welltolerated VT with localized forms of the disease anddrug-refractory or incessant VT or frequent ICDdischarges. In the latter, catheter ablation can playan important role as a palliative or an adjunctivetreatment option for reduction or suppression of theVT.
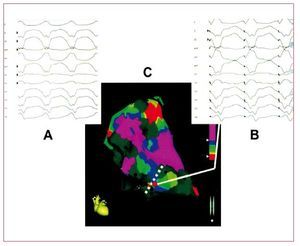
VT in ARVD is the result of a scar-relatedmacro-reentry circuit, similar to that observed in the post-myocardial infarction setting. RV voltagemapping catheter ablation, using both conventionalor electroanatomical mapping techniques, can offergood acute results (Figure 4).42 However, due to theprogression of the disease, VA recurrences from newarrhythmogenic substrates are frequent. Wichter etal43 reported their experience with catheter ablationin 30 patients with ARVD. Favorable acute resultswith complete suppression of drug-refractory VTwere achieved in 22 patients (73%). However,the long-term results were less satisfactory, with18 patients (60%) suffering from VT recurrence.Of note, the majority of late VT recurrences(>1 year) in this study showed a different QRScomplex morphology compared with the target VT,suggesting that new arrhythmogenic substrates maydevelop during long-term progression of the disease.The same results were reported by other authors.44,45 Subepicardial substrate has been also considered forVT recurrences.45

Figure 4. An example of the combinedused of voltage and pace mapping ina patient with arrhythmogenic rightventricular dysplasia. A: the 12 lead ECGwith the clinical ventricular tachycardia.B: the 12 lead ECG during pace mappingfrom the catheter tip (red dot at sitesalong the scar border). C: a near identicalECG morphology match with the clinicalventricular tachycardia, indicating thissite as the potentially critical exit zoneof a macro reentry circuit. Modified fromArruda M et al41 with permission.
Implantable Cardioverter-Defibrillator
In patients with ARVD, ICD therapy improvesthe long-term prognosis and survival when appliedin selected high-risk populations and as secondaryprevention (Figure 5).45-47 However, criteria for theoptimal selection of patients who will benefit fromICD implantation for primary prevention have not yet been defined. In the future, it is likely thatgenetics will play a more important role in decision-making.46,47

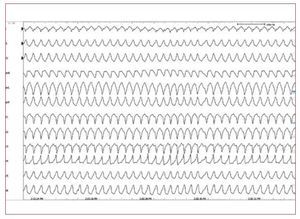
Figure 5. A stored intracardiacelectrogram of an implantablecardioverter-defibrillator showing fast VTwith appropriate defibrillation in a youngmale with arrhythmogenic right ventriculardysplasia.
Of note, ICD implantation in patients withARVD may lead to complications more frequentlythan in other diseases requiring an ICD. A commoncomplication is related to the progression ofmyocardial atrophy and subsequent replacement byfat at the site of lead implantation. This results in aloss of the sensing function in the right ventriculardefibrillation lead, with a concomitant need for leadrevision. Therefore, when deciding on whether to useICD therapy in ARVD, the potential benefits shouldbe weighed against the risk of complications.
When the disease has progressed to rightventricular or biventricular failure, treatmentconsists of the current therapy for heart failure,including diuretics, beta-blockers, angiotensin-converting enzyme inhibitors, and anticoagulants.In the case of intractable right heart failure, cardiactransplantation may be the only alternative.
BRUGADA SYNDROME
Introduction
Brugada syndrome (BS) is a genetic disease whichcauses cardiac arrhythmias and is characterizedby the occurrence of SCD in young individuals without evidence of structural heart disease. Dueto the macroscopic absence of structural heartabnormalities, BS is classified as a "primary electricaldisease" or cardiac channelopathy. BS is related tostructural and functional abnormalities in the sodiumchannel. Different pathophysiological findingssuggest that the electrical disorders associated withBS are mainly located in the RV and particularly inthe RVOT.
Patients with BS present an ECG patterncharacterized by ST segment elevation in the rightprecordial leads (V1-V3)48 and an incomplete orcomplete RBBB. SCD is caused by polymorphic VTand/or ventricular fibrillation (VF).49 BS is thoughtto be responsible for 4% to 12% of all SCD and forup to 20% of SCD in subjects without structuralheart disease.50,51 Since the ECG is dynamic and often concealed, it is difficult to estimatethe real prevalence of the disease in the generalpopulation.51,52 The prevalence of BS is estimatedat 1-5 per 10 000 inhabitants worldwide. It is lessfrequent in western countries and higher (>5 per 10 000) in Southeast Asia, especially in Thailandand the Philippines where BS is considered tobe the major cause of natural death in youngindividuals.53,54
Genetic Aspects
The first mutation related to the syndrome wasdescribed in 1998 by Chen et al55 and was identifiedin the gene SCN5A that encodes the a-subunitof the cardiac sodium channel.55-57 Mutations in the SCN5A gene are presently found in 18%-30%of patients with BS. Inheritance in BS occurs viaan autosomal dominant mode of transmission.To date, 293 other mutations have been foundin the same gene. In 2002, Weiss et al describeda second locus on chromosome 3, which was notlinked to SCN5A: the gene identified was theglycerol-3-phosphate dehydrogenase 1-like (GPD-1L) gene.58 Mutations in the GPD-1L gene, whichcodes an enzyme regulating the trafficking of thecardiac sodium channel to the cell surface, have been shown to reduce inward sodium currents byapproximately 50%.59 Recently, mutations in theCACNA1c and CACNB2b genes,60 which code forcalcium channels, and in the KCNE3 gene,61 whichcodes for a subunit regulating the Ito potassiumcurrent, have also been shown to be responsible forthe BS phenotype.
A BS ECG is not the only phenotype linked tomutations in the SCN5A gene.62 Recent evidenceindicates a lot of overlap in clinical presentation("overlapping syndromes"). The most frequentlyreported overlap is the concomitant presentationof BS and cardiac conduction disease (Lev-Lenegre disease, sick sinus node syndrome). Theoverlap between the LQT3 and BS phenotypeshas also been reported in several cases.63 In recent years, the association of atrial fibrillationwith known sodium channelopathies such as BS,progressive cardiac conduction disease, LQT3 andshort QT syndromes has been also reported.64-66 Interestingly, atrial fibrillation has been shownto be potentially the first phenotypic expressionof a latent form of BS which manifests only yearslater.67
Pathophysiology
Two main hypotheses have been proposed toexplain the pathophysiologic mechanisms ofECG abnormalities and susceptibility to VAs inpatients with BS.51 In 1999, the theory of "impairedrepolarization" was proposed.68 This theory wasbased on a non-homogeneous expression ofthe transient outward potassium current (Ito) between epicardium and endocardium. Ito, whichis responsible for the early repolarization phaseduring action potential (AP), is more stronglyexpressed in the epicardium. In the presenceof a loss or reduced function of the sodiumchannels, a "spike and dome" AP shape arises.The effect on the action potential will be moreevident in the epicardium layer, where the Ito is stronger. Thus, a discrepancy in the AP shapebetween endocardium and epicardium will resultin the surface BS ECG patterns. Abnormalitiesin the surface ECG will be proportional to thediscrepancy in the AP, creating slight ECGalterations (saddle-back ST elevation, J point <0.2 mv) or marked ECG alterations (covetype, J point >0.2 mv) according to the degreeof sodium channel damage and the different Itoexpression. The conduction of the AP dome fromsites where it is maintained to sites where it is lostprovokes local re-excitation via a phase 2 reentrymechanism with a closely coupled extrasystoleoccurring during the vulnerable period. ThesePVC may then trigger malignant VAs.
The second theory, called "depolarizationtheory," 69 is based on the presence of conductiondelay in the RVOT. According to this theory,the AP shape is maintained, but the RVOT AP isdelayed with respect to the RV AP. These AP timingdifferences in adjacent areas of myocardium maybe a source of reentry circuits triggered by PVCoriginating in the border zone between early anddelayed depolarization.A third theory, based on the abnormal expressionof the neural crest on myocardial development ofthe RVOT and surrounding structures, has beenrecently proposed by Elizari et al.70 During embryo-genesis, the cardiac neural crest plays a critical rolein morphogenesis of the RVOT, which comprises thefree wall and the aorto-pulmonary septum, and thegreat arteries. A population of cardiac neural crestcells migrates toward the arterial pole of embryonicheart leading to myocardial cell proliferation,differentiation and RVOT myocardialization. Asecond population of migratory cardiac neural crestcells enters the heart via the venous pole and plays acrucial role in the formation of the AV node, the Hisbundle, the beginning of the bundle branches, andatrial tissue.
Consequently, abnormalities in right heartformation related to the cresta neuralis fromoutside the heart could explain all of the electricalabnormalities (supraventricular arrhythmias,conduction disturbances, ventricular arrhythmias)potentially manifesting in patients with BS and theoverlapping syndromes.
In particular, a non-homogeneous expression ofconnexins, molecules which are highly expressedby neural crest cells, may be responsible for theerroneous migration of neural crest cells towardthe embryonic heart. Mistiming of neural crest cellmigration due to a connexins malfunction may havea deleterious effect on cardiac tissue remodelingof RV. Thus, abnormal RVOT myocardializationwhich depends on neural crest cell migrationmight explain the repolarization heterogeneitiesunderlying the BS phenotype. This modelhypothesizes an unequal distribution of repolarizingforces. According to this theory, repolarizationgradients causing ST-segment elevation occur notonly between the epicardium and endocardium(because of higher Ito expression in epicardiumthan in endocardium) but also between RVOT andnormal surrounding structures.
Clinical Presentation
The clinical presentation in patients with BScan range from a complete lack of symptoms tosudden death. Syncope, seizures, palpitations, andnocturnal agonal respiration have all been described as being the first symptom at presentation. Up to20% of patients in Western countries and up to30% in Japan have concomitant supraventriculartachycardias,66 most frequently atrial fibrillation,which can also be the first manifestation of thedisease.67
BS is mainly considered an arrhythmogenicdisease of adult males (80%), with a reported meanage at sudden death of 40 years.71 Clinical studiessuggest that the male hormone, testosterone, maybe responsible for the gender differences observed inthe prevalence of BS.72-74
ECG and Clinical Characteristics
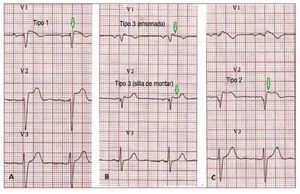
According to the Second Consensus Conferenceon BS, 3 ECG repolarization patterns are currentlyrecognized, (Figure 6). Type 1, which is the onlyone diagnostic of BS, is characterized by a covedST-segment elevation ≥2 mm (0.2 mV) followedby a negative or flat T wave. The type 2 ECGrepolarization pattern is characterized by ST-segment elevation, which has a "saddleback"appearance with a high takeoff ST-segmentelevation of ≥2 mm and either positive or biphasicT wave. Type 3 has either a saddleback or covedappearance with an ST-segment elevation of <1mm. Type 2 and 3 are not diagnostic for BS, but thediagnosis of BS is also considered positive when atype 2 or 3 pattern is observed in >1 right precordiallead under baseline conditions and conversion tothe diagnostic type 1 pattern occurs after sodiumchannel blocker administration.75 ECG recordings from V1 and V2 leads at higher (3rd and 2nd)intercostal spaces increase the sensitivity andspecificity of the ECG diagnosis for the Brugadaphenotype.76,77

Figure 6. A: a spontaneous type 1Brugada syndrome ECG. B: a "saddle-back" and a "coved" type 3. C: a type2 Brugada syndrome pattern. Only typeI should be considered diagnostic ofBrugada syndrome.
Consequently, according to the SecondConsensus Conference criteria, BS is diagnosedwhen a type 1 ST-segment elevation is observed in >1 right precordial leads (V1-V3) in the presenceor absence of a sodium channel-blocking agentand in conjunction with one of the followingevents: documented ventricular fibrillation, (self-terminating) polymorphic VT, a family historyof SCD under the age of 45 years, presence ofcoved-type ECG in family members, inducibilityof VT with programmed electrical stimulation orsyncope. Patients displaying the characteristiccoved-type ECG pattern without further clinicalcriteria should be considered as having a BrugadaECG pattern and not a BS. Very recently, we haveshown that a type I ECG in one precordial lead issufficient for the diagnosis.78
All the 3 patterns described above may beobserved spontaneously in serial ECG tracings fromthe same patient, as well as "pseudo-normalization" of the ECG.79 In clinical practice, these ECGfluctuations can make it quite difficult to identifyindividuals affected by BS and at risk for SCD. Inaddition to sodium channel blockers, many agentsand conditions are reported to unmask a type 1BS ECG phenotype, including body temperature,changes in autonomic tone and drugs affecting ionchannel function, such as calcium-channel blockers,beta-blockers, antiarrhythmic drugs, psychotropic drugs and alcohol or cocaine toxicity.75 Recently,Amin et al showed that exercise, especially duringthe recovery phase, is also able to unmask a type 1BS-ECG or to increase the precordial peak J pointelevation.80
Diagnostic Tools: the Class I AntiarrhythmicDrugs Challenge
Given that the ECG is dynamic and that thecharacteristic ECG hallmark may thus be concealed,drug challenge with sodium channel blockers thatincrease the sodium channel dysfunction has beenproposed as a useful tool for the diagnosis of BS.Currently, ajmaline, administered as an intra-venousdose of 1 mg/kg over 5 minutes, represents the firstchoice due to its high sensitivity and specificityin identifying gene carriers (80% and 94.5%respectively).81 Additionally, the short half-life andbrief duration of its electrophysiological effectsrender it safer than other antiarrhythmic drugs.
Risk Stratification
The presence of a spontaneous right precordial STsegment elevation (coved type), history of clinicalsymptoms (syncope, aborted sudden death), andmale sex are risk factors for malignant clinical eventsin patients with BS.49,82-84
There is general agreement that survivors of cardiacarrest have a high risk of recurrent life-threateningarrhythmic events and should therefore receive anICD.34,37 Previous syncope may be reported in up to23% of patients who experience an episode of cardiacarrest.84 Neurally mediated syncope has also recentlybeen associated with BS, but its implications forprognosis and risk stratification are still unknown.85 Males have consistently shown a tendency to presentmore arrhythmic events in all studies, and beingmale was identified as an independent predictorfor poorer outcome in a recent meta-analysis.86 The best management approach in asymptomaticpatients and whether electrophysiological studiesare useful or not in predicting adverse events is stillcontroversial.84,87,88
Treatment
The only proven therapy to prevent sudden deathin symptomatic Brugada patients is an ICD.50,89-90 Quinidine can be used as a pharmacologicaltherapeutic option. Its use might be particularlyuseful in patients with an ICD and multiple shocks42 and as an initial therapeutic option in youngpatients at high risk for malignant arrhythmias.However, real scientific data on its efficacy are stilllacking.
RIGHT VENTRICULAR OUTFLOW TRACTVENTRICULAR TACHYCARDIA
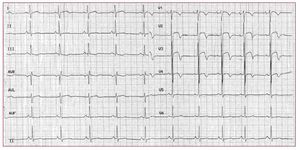
RVOT VTs are the most common form ofidiopathic VTs.91-92 In most cases (70%-80%),93 RVOT VT does actually originate in the RVOT;however, other origins are possible, includingthe septum, the left ventricular outflow tract, thepulmonary artery, the aortic sinus of Valsalva, thearea near the His bundle, and the epicardial surfaceof the ventricles.94 The characteristic morphology ofthe RVOT-VT is a wide QRS complex tachycardiawith LBBB morphology and an inferior axis (Figure 7). Ventricular tachycardia is monomorphic andgenerally not familial.

Figure 7. The 12 lead ECG is from a31-year-old female and shows a sustainedventricular tachycardia originating fromthe right ventricular outflow tract (leftbundle branch block-like QRS complexesand inferior axis). Few radiofrequencyapplications around 1 cm beneath thepulmonary valve on the free wall siderendered the ventricular tachycardiauninducible.
Clinical presentation of RVOT-VT is variable andsymptoms usually occur from the third to the fifthdecade of life.95 RVOT-VT occurs more frequentlyin women.96 Two phenotypic forms of RVOT-VTare known: exercise or stress-induced sustainedVT and nonsustained, repetitive monomorphic VTat rest. Both forms are characterized by adenosinesensitivity. Nonsustained VT, which usually occursas repetitive runs of monomorphic VT, is frequentcomprising 60%-92% of reported series. Mostpatients present with palpitations or presyncope,and rarely present with syncope. Exercise oremotional stress usually precipitates the tachycardia.Resting ECGs in these patients show no identifiableabnormalities. Echocardiogram and coronaryangiography are normal in most of the patients.97 The MRI may show abnormalities in up to 70% ofpatients, including focal thinning, diminished wallthickening, and abnormal wall motion.98 However,the real meaning of these "abnormalities" is notknown.
RVOT-VT shows a benign course, suggestingthat, in the majority of cases, this arrhythmia is nota first form of an occult cardiomyopathy. Gaita etal reported that 61 patients with RVOT ventricularextrasystoles were contacted 15 years after the initialvisit but found that no patients had died of SCD ordeveloped ARVD in the study.99
However, some authors have reported that "malignant arrhythmias" such as ventricularfibrillation and/or polymorphic VTs, are sometimesassociated with idiopathic VT or PVC originating inthe RVOT.100,101 Patients potentially at high risk forthis "malignant" variant appear to be those with: a)a history of syncope; b) very fast VT (heart rate >230bpm); c) PVC with a short coupling interval; and d) long average QRS duration of the initiating PVCoriginating from the RVOT.101 Catheter ablationshould be strongly considered for patients with thelatter characteristics.
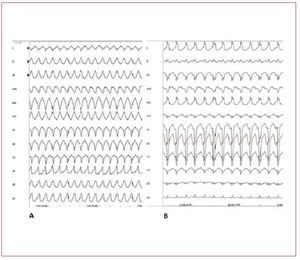
In the differential diagnosis ARVD should bestrongly suspected and investigated. The first feature that can differentiate RVOT-VT fromARVD is that RVOT-VT always presents withLBBB morphology and inferior axis. VT in ARVDusually has a LBBB morphology but the QRScomplex can have different orientations in thefrontal axis. The differential diagnosis of RVOTVT has also to exclude tachycardia associated withatriofascicular fibers, atrioventricular reentranttachycardia using a right-sided accessory pathway,and VT occurring in patients after repair oftetralogy of Fallot (Figure 8).

Figure 8. A: sustained ventricular tachycardia (VT) originating from the right ventricular outflow tract. B: sustained VT due to atriofascicular fibers. Although both VTs present with wide QRS complex and left bundle branch block, the QRS axis in the frontal plane is different, reflecting 2 different mechanisms and origins. In case A, the VT has a focal source and the ventricular myocardial mass is activated from top to bottom. In case B, the VT is dependent from a reentry circuit and the ventricular myocardial mass is activated in the opposite direction, from the bottom to the top, thus creating a different direction of the QRS axis in the frontal plane. Atriofascicular fibers are uncommon but a very distinct form of accessory pathways. These accessory pathways arise from the right atrium at the parietal tricuspid ring and terminate in or near the distal part of the right bundle branch. They exhibit slow and exclusively antegrade decremental conduction. Because of the right sidelocation of these pathways, preexcitedQRS complexes show a left bundle branchblock-like morphology.
Mechanism of Right Ventricular Outflow TractVentricular Tachycardia
Triggered activity is considered to be themechanism underlying RVOT-VT, due tocyclic adenosine monophosphate (cAMP)-mediated delayed afterdepolarizations.102,103 An intracellular calcium overload is present inpatients suffering from RVOT-VT. Cytosoliccalcium overload enhances the function of theNa+/Ca++ exchanger, which leads to increasinginward current and delayed afterdepolarization.Adenosine is effective in terminating RVOT-VTbecause of its ability to lower cAMP, which hasa crucial role in regulating intracellular calcium.The tachycardia may be inducible by programmedextrastimuli, by burst pacing the ventricle oratrium or by infusion of isoproterenol duringelectrophysiological study.
Treatment
The management strategy for RVOT-VTincludes antiarrhythmic drugs and catheterablation. If symptoms are very mild or infrequent,no treatment is necessary. RVOT-VT can bearrested acutely by vagal manoeuvres, carotidsinus massage, intravenous adenosine (6-18 mg),and intravenous verapamil (5-10 mg). Efficacyof antiarrhythmic drugs (beta-blockers, calciumchannel blockers) in patients with RVOT-VTis around 25%-50%. Considering the long-termsuccess rate (>90%) and the low incidence ofmajor complications (<1%),93,97 catheter ablation is becoming the first-line therapy in patients withRVOT-VT.
CONGENITAL HEART DEFECTS (TETRALOGYOF FALLOT)
Rhythm disturbances may be observed in thenatural history of congenital heart defects (CHD),as well as after open heart surgery.104 An increasing number of patients with CHD are reachingadulthood, due to the success of corrective surgicalprocedures.105 About 40%-50% of adults with CHDwill experience some type of arrhythmia. The highincidence of cardiac arrhythmias in the adult withCHD can be caused by abnormal pressure/volumechanges and more often by re-entrant circuitscreated by septal patches and suture lines.105,106 Supraventricular tachycardias, such as atrial flutter and/or atrial fibrillation, are common in CHD(Figure 9).

Figure 9. The 12 lead ECG shows an atrialflutter in a 42-year-old female who hadundergone tetralogy of Fallot correction31 years earlier. The patient was admittedto our center because of palpitations. Atypical counterclockwise right atrial flutter(cavo-tricuspid isthmus dependent) wasfound during an electrophysiology studyand treated with radiofrequency ablation.A 12 lead ECG after the ablation stillshowed right bundle branch block andright axis deviation, reflecting the rightheart dysfunction.
VT is seen in a smaller number of patients,most notably in those with tetralogy of Fallot(TOF). This condition consists of four features:subpulmonary infundibular stenosis, ventricularseptal defect, overriding aorta, and right ventricularhypertrophy. The surgical procedure includes a rightventriculotomy and ventricular septal defect patchrepair. These lesions cause the arrhythmia substrate.The ventricular septal defect patch repair providesa fixed anatomic obstacle around which re-entrantarrhythmias may occur.
After repair of TOF, sustained VT has a prevalenceof 4%-7%107 and is usually a RVOT reentranttachycardia with LBBB morphology. NonsustainedVAs are present in up to 60% of Holter recordings.
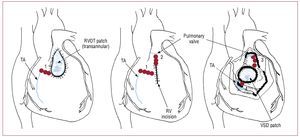
In these patients, there is a persistent risk of lateSCD, with an estimated incidence rate of 0.5%-8.3%.108,109 Older age at initial repair, moderate or severe pulmonary regurgitation, a history ofsustained VT, moderate or severe ventriculardysfunction and a QRS duration ≥180 ms arepredictive of risk of SCD.109 Nonsustained VTin patients with TOF has shown no predictivevalue for subsequent sustained VT or SCD.108 The patients usually present with palpitations, less often with syncope or presyncope. Antiarrhythmic drugssuch as amiodarone and sotalol are usually used inthese patients as first-line therapy. In patients withdrug-refractory symptomatic VT, radiofrequencyablation can be performed. Conventional andelectroanatomical mapping are used to identify thereentry circuit. Electroanatomical mapping may becrucial for success, especially in poorly tolerated VTs.An isthmus between an area of anterior wall RVOTscar/patch and the tricuspid annulus (Figure 10) hasbeen shown to be the main cause of VTs (75%) insymptomatic patients undergoing radiofrequencyablation after repair of TOF. Long-term successcan be achieved in about 90% of patients.110 An ICDis indicated in patients with aborted SCD and VTwhich cannot be ablated.

Figure 10.Localization of anatomic boundaries (blue line) for right ventricular tachycardia after repair of cardiac heart defect and the resulting anatomic isthmuses (red lines, numbers from 1 to 4). RV indicates right ventricle; RVOT, right ventricular outflow tract; TA, tricuspid annulus; VSD, ventricular septal defect. Modified from Zeppenfeld K et al107 with permission.
Right Ventricle, ECG Abnormalities, and Sudden Cardiac Death in Athletes
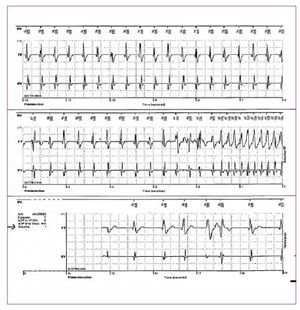
It has been estimated that VAs in right ventriculardisease such as BS and ARVD account for 10%-30%of SCD in young adults in the general population. Thispercentage is even higher in young athletes.2 Spain hasrecently suffered a plague of sudden deaths amongyoung national soccer players affected by ARVD.It has been reported that 20-35 young athletes die suddenly in Spain each year. In young, otherwiseapparently healthy subjects, SCD is always a tragicevent and can be even crueller when photographedfor viewing by millions of people. Pre-participationscreening has been shown to decrease SCD amongathletes.111 Abnormal 12-lead ECG in trainedindividuals are frequent. Common and training-related abnormalities include sinus bradycardia,first degree and Wenckebach heart blocks,incomplete RBBB, left ventricular hypertrophy andan early repolarization pattern. These physiologicabnormalities reflect the remodelling of the heartand the change in autonomic balance in individualswho train regularly and intensely. However, in asmall percentage of cases (8%),112 ECG changes maybe marked and diffuse. A careful interpretation andinvestigation (Figure 11) is required in these casesto rule out life-threatening disease, especially inpresence of syncopes and/or a family history of SCD.Cardiac genetic disorders (ARVD, BS, hypertrophiccardiomyopathy, long-short QT syndrome) havevarying levels of environmental contribution that can worsen or even unmask the phenotypicexpression of the disease. In patients with ARVD,exercise accelerates the genetically determineddamage of mechanical contacts.113,114 Exercise, especially during the recovery phase, is also able tounmask a type 1 BS-ECG and consequently to exposethe subjects to life-threatening conditions.81,115-117 In general, patients with ARVD and BS should beadvised against strenuous exercise and vigoroustraining and should be excluded from participationin competitive or professional athletic sports.

Figure 11. The ECG is from an asymptomatic 17-year-old competitive soccer player at rest without evidence of left ventricular hypertrophy or other structural abnormalities on echocardiography. The ECG shows marked repolarization abnormalities, including ST-segment elevation and T wave inversion in precordial lead V1-V4. With this ECG pattern the exclusion of life-threatening cardiac conditions such as hypertrophic cardiomyopathy, arrhythmogenic right ventricular dysplasia,dilated cardiomyopathy, aortic valvestenosis, and channelopathies (Brugadasyndrome, long and short QT syndrome)is mandatory.
ICD implantation should be seriously consideredafter a careful risk stratification. Debate exists asto whether athletes who have received an ICD may or may not continue competitive sports. Accordingto the Bethesda guidelines,118 patients with an ICDcan participate only in "class IA" activities, suchas bowling or golf. Conversely, more recent datashowed that, although shocks during sports arereported frequently, significant adverse events arerare.119 Therefore, physician recommendationsregarding participation in sports still vary widely.Several theoretical and practical reasons existto restrict athletes with ICDs from participationin sports but data actually proving sports to bedangerous for all ICD patients are not available andan informed decision should probably lie with theindividual athlete.
ABBREVIATIONS
ARVD: arrhythmogenic right ventricularcardiomyopathy/dysplasia
BS: Brugada syndrome
CHD: congenital heart defects
PVC: premature ventricular contractions
RV: right ventricle
RVOT: right ventricular outflow tract
SCD: sudden cardiac death
VA: ventricular arrhythmias
Correspondence: L. Capulzini MD,
Heart Rhythm Management Center. UZ Brussel-VUB,Laarbeeklaan 101, 1090 Brussels. Belgium
E-mail: luciomassimo@hotmail.com