.
Insulin resistance, hyperinsulinemia and vascular disease: defining the problemMacro- and microvascular complications are both main causes of morbidity and mortality in type 1 and type 2 diabetes,1 but macrovascular complications have an increased incidence even before the onset of type 2 diabetes.2 While high blood glucose3 and glucose-induced protein and lipid modifications—the advanced glycation endproducts4—can be triggers of both macro- and microvascular disease once diabetes (both type 1 and type 2) has occurred, factors causing macrovascular disease in the setting of the metabolic syndrome and prediabetes have long been debated. Certainly in diabetes, and probably also in the setting of the metabolic syndrome,5, 6, 7, 8 atherosclerotic vascular and coronary heart disease occur over and above the clustering of other accompanying risk factors, such as hypertriglyceridemia, low levels of high-density lipoproteins and hypertension. Insulin resistance before the onset of diabetes is, by definition, characterized by hyperinsulinemia, and this has long been speculated to be causally linked to vascular disease.9, 10, 11, 12 In this brief review we will address the biological plausibility and the evidence for hyperinsulinemia to be a causal mechanism in the development of atherosclerosis preceding and following the onset of type 2 diabetes.
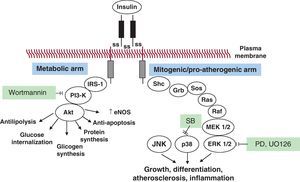
Selective insulin resistance and compensatory hyperinsulinemia: pathophysiologyOriginally, Reaven et al. defined the insulin resistance syndrome as a cluster of cardiovascular risk factors, including glucose intolerance, dyslipidemia, and hypertension, associated with enhanced cardiovascular disease.13 They described the metabolic syndrome as a clinical condition characterized by insulin resistance, impaired fasting plasma glucose, obesity, dyslipidemia, and hypertension.13 Later, two definitions of the metabolic syndrome have been proposed, one by the National Cholesterol Education Program Adult Treatment Panel III14 and the other by the World Health Organization.15 Here, insulin resistance—as measured by the reference method of the hyperinsulinemic euglycemic clamp or by surrogate methods, such as the frequently sampled intravenous glucose tolerance test, insulin suppression test, or HOMA index—can be demonstrated in up to 76% of subjects,16 and is accompanied by compensatory hyperinsulinemia.16 Although molecular mechanisms of insulin resistance are still incompletely understood, abnormalities in insulin signaling have been described.17 In peripheral tissues, including the skeletal muscle and the liver, in normal conditions insulin initiates its action by binding its specific cell surface receptor, ie, insulin receptor (IR), which is a heterotetrameric protein consisting of 2 extracellular α-subunits and 2 transmembrane β-subunits connected by disulfide bridges. Insulin binding to the extracellular α-subunit induces conformational changes of the IR, which in turn causes the dimerization of adjacent receptors and the activation of the tyrosine kinase domain of the intracellular part of the β-subunit. Once the tyrosine kinase activity of IR is initiated, it promotes the autophosphorylation of the β-subunit itself and the rapid phosphorylation of so-called “docking proteins”, such as IR substrates (IRS)-1, -2, -3, and -4, and several other proteins, including collagen homology proteins (shc) and src homology 2 (SH2), that in turn activate multiple intracellular signaling intermediates (Figure 1). Therefore, IRS, shc, and SH2 proteins play important regulatory roles in the insulin signaling cascade. In their phosphorylated form, these proteins become points of anchoring for intracellular proteins containing complementary SH2 domains. In particular, the interaction between IRS-1 proteins and phosphatidylinositol (PI) 3-kinase determines the activation of Akt (also known as protein kinase B), which is critical in the mechanism of insulin action on GLUT-4 translocation, glucose transport, and the activation of nitric oxide (NO) synthase (“metabolic signal pathway”). In contrast, nonmetabolic, proliferative, mitogenic, pro-inflammatory effects of insulin are mediated by the activation of Ras (mostly through shc and, to a lesser degree, IRS proteins), Raf, and mitogen-activated protein kinases (MAPK) (“growth signaling pathway”).18 In insulin-resistant animals and in vitro models a reduced activation of insulin signaling via the IRS-1/PI3-kinase pathway can be demonstrated, resulting in diminished glucose uptake, reduced NO synthesis, and reduced glucose utilization in insulin target tissues. The same decrease in glucose transport is sensed at the level of pancreatic beta cells, and induces a compensatory increase in insulin secretion. At the same time, however, the MAPK-mediated insulin pathway remains unaffected.19 One can easily understand that this selective imbalance of the two signal transduction pathways in such conditions of hyperinsulinemia may lead to an excessive proliferative/growth-promoting signal, at the same time allowing the maintenance of normal glucose transport and glucose homeostasis. Compensatory hyperinsulinemia stimulates various proliferative and pro-atherogenic events in vascular smooth muscle and endothelial cells. Such effects include the increased production of plasminogen activator inhibitor type-1 (PAI-1), endothelin, proinflammatory cytokines and the increased surface expression of adhesion molecules.19, 20, 21, 22

Figure 1. The insulin signaling pathway and its impairment in insulin resistance. Upon binding to its tyrosine-kinase receptor, insulin induces receptor dimerization, and activation of a cascade of phosphorylation events, producing two classes of effects: a) “metabolic” effects, promoting glucose transport, glycogen and protein synthesis, inhibition of lipolyis, protection from apoptosis, and the release of nitric oxide (broadly described as “anti-inflammatory” effects), and b) growth- and differentiation-promoting effects, which lead to promotion of inflammation and atherogenesis (ie, mitogenic, pro-inflammatory insulin signaling). Akt, protein kinase B (PKB); eNOS, endothelial nitric oxide synthase; ERK, extracellular receptor kinase; IRS-1, insulin substrate receptor-1; JNK, c-Jun NH2-1 terminal kinase; MEK, mitogen-activated protein kinase/extracellular receptor kinase; p38, p38 mitogen-activated protein kinase; PD (PD98059) and UO126, extracellular receptor kinase 1/2 inhibitors; PI3-Kinase, phosphatidylinositol(PI)3-kinase; wortmannin, PI3-kinase inhibitor.
Insulin has an important role in the maintenance of blood vessel homeostasis through the activation of endothelium-derived NO. Insulin increases endothelial NO production by activating NOS-III (endothelial NOS) by rapid posttranslational mechanisms, which are mediated by the PI3K/Akt signaling pathway.23 In insulin resistance states the PI3K/Akt pathway is selectively inhibited, and this leads to endothelial dysfunction, with a consequent increase in vascular tone and hypertension, increased interaction between endothelial cells and leukocytes, and a prothrombotic state. This “selective” insulin resistance has been shown in skeletal muscle from obese people and patients with type 2 diabetes,24 and in the vasculature and the myocardium of obese Zucker rats. Here, the normal physiological anti-atherogenic effects of insulin, due largely to its capacity to increase NO production, are turned into pro-atherogenic effects.25
A vicious circle between hyperinsulinemia and insulin resistanceHigh plasma concentrations of insulin in conditions of insulin resistance also can trigger a vicious circle that further increases insulin resistance26 through a suppression of the effects mediated by the PI3K/AKT/NO axis, which may unbalance the system through the net promotion of effects related to MAPK activation. Since insulin triggers a series of biological effects through the binding and activation of its receptor (IR), endowed with tyrosine-kinase activity on specific substrates including IRS -1 and -2,27 mice with a targeted deletion of IRS-1 and IRS-2 genes have a phenotype of insulin resistance.28
Animal models of hyperinsulinemia, such as the ob/ob mice and the obese Zucker rats, have low levels of IRS-1 and IRS-2 proteins in the liver.29, 30 These models are characterized by insulin resistance and a reduced function of the IR/IRS-1/PI3K/AKT axis in the liver and the skeletal muscle. It has been shown that brief in vitro incubations of myoblasts with high concentrations of insulin determine a PI3K-mediated reduction of IRS-1 protein expression and a desensitization of insulin signal transduction mechanisms.9 Finally, a prolonged exposure of cultured myoblasts to high concentrations of insulin is associated with a reduction of the IR/IRS-1/PI3K/AKT axis activity.31 We have demonstrated that prolonged exposure of human umbilical vein endothelial cells to high insulin levels induces a downregulation of the PI3K(AKT/eNOS axis, paralleled by increased expression of vascular cell adhesion molecule 1 (VCAM-1).32 However, the molecular mechanisms through which hyperinsulinemia begets or worsens insulin resistance are still largely unknown.
Hyperinsulinemia and vascular disease: Evidence from the benchAnimal experiments33, 34 and several in vitro studies provided evidence for the biological plausibility of the hypothesis according to which high concentrations of insulin are pro-atherogenic. A link between coronary artery disease and high insulin concentrations was first proposed in the late 1960's10 and subsequently confirmed (for a review, see Reddy et al.35). In vitro, insulin has been shown to stimulate the proliferation and migration of arterial smooth muscle cells in tissue culture preparations21 and to induce monocyte adhesion by increasing the expression of VCAM-1 in endothelial cells.22, 36, 37 VCAM-1 is probably the adhesion molecule most relevant to the development of atherosclerosis.38 Such increased expression in the presence of insulin occurs in a system where insulin may still increase NO bioavailability, which would normally inhibit endothelial activation and atherogenesis.39 Therefore these findings mean that a net effect of high insulin concentrations on endothelial cells is mostly a pro-inflammatory phenotype. We have also shown that these effects can be potentiated by the PI-3-kinase inhibitor wortmannin,22 leading us to postulate that they may be further amplified in conditions of insulin resistance mimicked by wortmannin. Because insulin's ability to induce endothelial activation (for which VCAM-1 expression is both a marker and a mediator) is a plausible explanation for macrovascular disease accompanying hyperinsulinemic conditions, we examined potential molecular mechanisms involved in this specific pattern of endothelial activation. Human umbilical vein endothelial cells were incubated with insulin (0-24h)±inhibitors of signaling pathways potentially involved. Incubation of endothelial cells with inhibitors of ERK1/2 failed to affect insulin-induced VCAM-1 expression. Conversely, the p38 MAPK inhibitors SB203580 and SB202190, the protein kinase C (PKC)-β isoform inhibitor LY379196, and—partially—the c-Jun NH2-terminal kinase inhibitor SP600127, all tested at concentrations around their IC50 for inhibition of substrate phosphorylation, decreased insulin effect on VCAM-1. Gene silencing of p38 MAPK by small interfering RNAs, which suppressed the expression of p38 MAPK, suppressed insulin-stimulated VCAM-1 expression.22, 36, 37 Treatment with insulin also led to activation of NF-κB.22, 36
In animals, long-term treatment with insulin has been shown to induce arterial lesions which are rich in lipids and to stimulate wall thickening.10 The mechanisms responsible for these lesions are increased cholesterol synthesis in the adipose tissue, an imbalanced ratio between receptors for low density lipoproteins and high density lipoproteins (with an increase of the former and a reduction of the latter), and increased low density lipoproteins binding to arterial smooth muscle cells.10 Insulin is also a growth factor able to promote angiogenesis and smooth muscle cell proliferation by activating the same pathways that are activated by IGFs.40 These insulin effects appear to be involved in the retinal neovascularization, playing therefore a key role in the pathophysiology of diabetic microangiopathy and—potentially—atherosclerotic plaque destabilization.41, 42, 43
Among the other potential mechanisms by which high levels of insulin favor atherosclerosis, endothelial dysfunction,44 and inhibition of macrophage apoptosis are probably also relevant.45 Endothelial dysfunction precedes and predicts macrovascular events. In healthy human subjects, the infusion of insulin, attaining pathophysiologically relevant insulin concentrations (>120pmol/L), can induce severe endothelial dysfunction in large arteries.44 The responsible mechanisms likely involve increased intracellular oxidative stress.46 In vitro studies have shown that insulin stimulates the production of endothelin, the activity of the sympathetic system, and sodium retention.47 Furthermore, insulin facilitates smooth muscle cell migration and proliferation, increases the production of extracellular matrix, and induces a pro-coagulant status,48 thus also possibly contributing to postangioplasty restenosis, more frequently observed in diabetic compared with nondiabetic patients.49
Hyperinsulinemia and cardiovascular disease: Evidence from the bedsideDespite the strong pathophysiology and experimental evidence for proatherogenic effects of hyperinsulinemia secondary to insulin resistance, patients with type 2 diabetes very often undergo insulin administration in order to normalize hyperglycemia, free fatty acids levels, and glycated hemoglobin. This treatment often implies insulin administration at very high doses (up to 100 or even 625 U/day),50 which causes the appearance of untoward effects, such as weight increase, inhibition of residual endogenous insulin secretion,51 and the overexpression of the MAPK pathway.19 However, because of the favorable effects of insulin on blood glucose and on high glucose-mediated deleterious effects of vascular function, the evidence for net deleterious effects of high doses of insulin in diabetes cannot be clear-cut. The DAI (Diabetes and Informatics Study Group, Italian Association of Diabetologists and Italian National Institute of Health) Study,52 a multicenter cohort study on the prevalence and incidence of cardiovascular events (myocardial infarction, cerebral thromboembolism, and peripheral amputations) in type 2 diabetic patients, showed that, compared with oral antidiabetic treatment (such as metformin, not entailing increased insulin secretion), insulin treatment was associated with a higher number of cardiovascular events in men and women with type 2 diabetes. In patients with type 2 diabetes, insulin therapy has been shown to independently increase the risk of foot ulcers,53 hypertension,54 and high ADP-dependent platelet aggregation.55 In the Framingham Heart Study, diabetic patients treated with insulin showed the highest incidence of morbidity and mortality for cardiovascular disease.56 In the First National Health and Nutrition Examination Survey, among 7381 observed patients, those with diabetes treated with insulin showed increased risk of all-cause death and death attributable to cardiovascular disease.57 In the Veterans Affairs Cooperative Study on Glycemic Control and Complications in Type II Diabetes, patients under intensive insulin therapy showed a 32% incidence of cardiovascular events compared with 21% in patients under standard insulin therapy.58 In the Atherosclerotic Risk in Communities study, patients on treatment with sulfonylureas (which also leads to increased insulin concentrations) had a relative risk for cardiovascular disease of 1.82, while patients on insulin therapy had a relative risk of 2.64.59 The Kumamoto study, where patients on insulin treatment did not show an increased risk of macrovascular disease, did not contribute substantially to addressing this question because the patients were hypoinsulinemic and nonobese.60 A recent study has shown that the mean amplitude of glycemic excursions from continuous glucose monitoring data correlated positively and independently with urinary 8-iso-prostaglandin F2α excretion, a marker of oxidative stress, in patients with poorly controlled diabetes on oral hypoglycemic agents.61 The authors did not find such associations in insulin-treated type 1 and type 2 diabetic patients, suggesting that insulin treatment itself inhibits oxidative stress in these patients. Yet, insulin effects on cellular homeostasis could also depend on insulin concentrations, since supra-physiological doses of insulin have been shown to induce generation of reactive oxygen species in vitro.62 Overall, exogenous insulin produces favorable (reduction of hyperglycemia) and adverse (promotion of atherogenesis) effects.63 This is a warning for a less extensive use of insulin in type 2 diabetes. In patients with blood glucose levels>300mg/dL, an initial insulin administration can decrease glucotoxicity50, 64, 65: after that, a reduction of insulin resistance by weight reduction, an increase in physical exercise, and the use of insulin sensitizers, such as metformin or the glitazones, would probably be a more rational choice to prevent cardiovascular complications in patients with type 2 diabetes. Of note, 5 large randomized trials of intensive glucose control vs standard therapy in type 2 diabetes have demonstrated no reduction in total or cardiovascular mortality58, 66, 67, 68, 69; in contrast, such reduction has been found in the EDIC Study70 in type 1 diabetes, where insulin resistance is not the primary problem and insulin treatment replaces a primary failure of insulin production by the pancreatic beta cells.
ConclusionsPathophysiological insulin concentrations increase the production of endothelin, proinflammatory cytokines, endothelial leukocyte adhesion molecules, and PAI-1, exerting overall vascular pro-inflammatory effects. Results of in vitro and in vivo studies point to a pathogenic role of pathophysiological and pharmacological insulin concentrations in vascular disease. Further research on the use of specific inhibitors of the MAPK and PKC pathways, as novel pharmacological agents addressing the pro-atherogenic insulin signaling, are warranted.
Conflicts of interestNone declared.
FundingThe original work of the authors here reported was supported by grants from the Istituto Nazionale per le Ricerche Cardiovascolari to Raffaele De Caterina.
Received 4 November 2011
Accepted 11 November 2011
Corresponding author: Istituto di Cardiologia, Università G. d’Annunzio-Chieti, C/o Ospedale SS. Annunziata, Via dei Vestini, 66013 Chieti, Italy. rdecater@unich.it