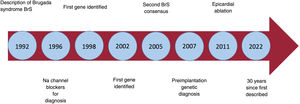
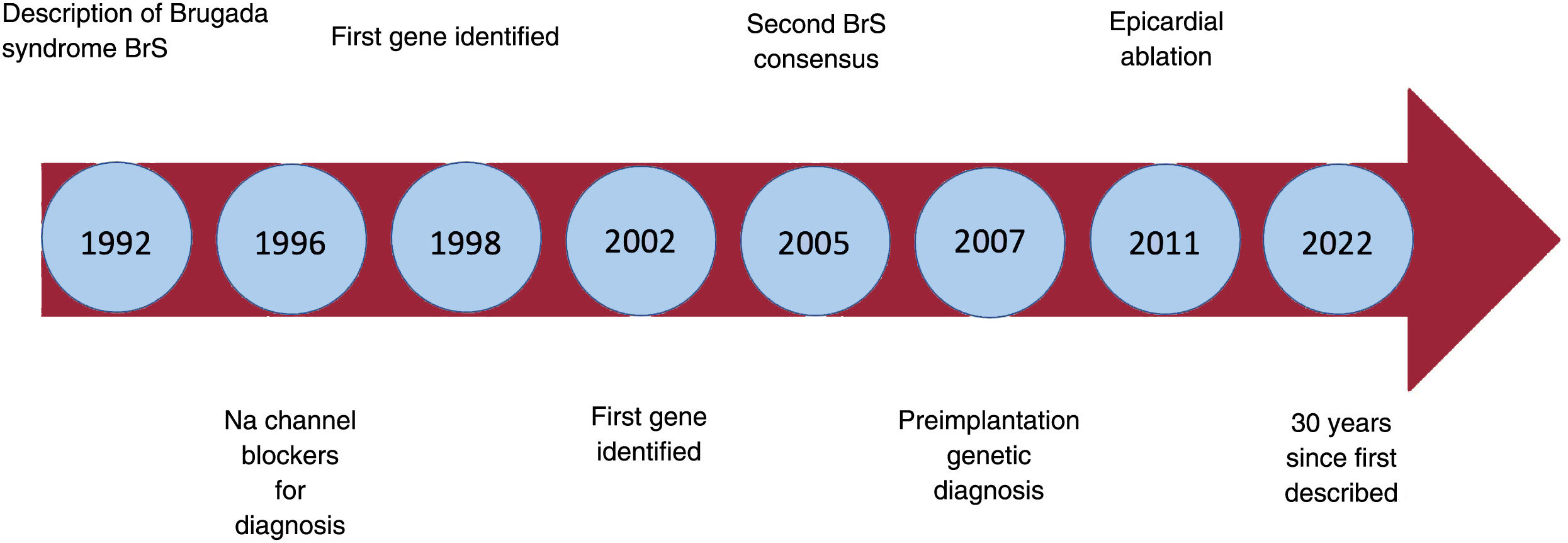
More than 30 years ago, on November 15, 1992, the Journal of the American College of Cardiology published an article titled “Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome. A multicenter report”.1 The article described 8 patients with a history of aborted sudden cardiac death (SCD) caused by ventricular fibrillation (VF). Despite extensive investigation, all attempts to identify the cause of the arrhythmias in these patients were unsuccessful. The 8 patients had a highly unusual electrocardiogram with ST-segment elevation in the right precordial leads and what appeared to be a right bundle branch block. The causes of the syndrome were unknown at the time, but it soon became clear that it was a purely electrical heart problem and that it must be hereditary. The very rapid VF indicated a problem of dispersion of short or normal refractory periods, in contrast to the relatively slower VF of long QT syndrome, in which the refractory periods are prolonged due to prolonged repolarization.1 Today we know that the electrocardiographic pattern of Brugada syndrome (BrS) is the final common pathway of various pathophysiological mechanisms, in some cases along with structural abnormalities, although in most cases, without.
It took 5 years to collect the data on the first 4 patients, presented in a poster at a conference of the North American Society of Pacing and Electrophysiology in 1991. After the presentation, due to international collaboration, a large number of potentially similar patients were compiled. Finally, 4 new patients with the same characteristics as the first 4 were selected. This spontaneous international collaboration (with no funding, protocols, or advisory committees) resulted in one of the most cited original publications in cardiology. Something the authors at first considered a curiosity became a true scientific revolution (figure 1).
 from its first description to the present day.")
With the description of this new syndrome, the ECG has proven itself as a simple, inexpensive, but highly valuable diagnostic tool: the diagnosis of BrS is based on an abnormal ECG. The only requirement for diagnosis is a “type 1” ECG, after exclusion of other possible causes (phenocopies).
BrS has once again shown the danger of classifying uncertain ECGs as normal variants. The ECG of BrS was, for years, considered a normal variant with no diagnostic or prognostic dignificance.2 It was a tough lesson to learn of our own naivety, faced with the evidence that these patients can have SCD. There have been efforts to structure the diagnosis of BrS using a points system.3 Unfortunately, this scoring system has no value in clinical practice, as up to 40% of patients with proven BrS would not score enough for the diagnosis.4
We define the electrocardiographic pattern as the presence of a type 1 pattern, either spontaneously or on a pharmacological test. In the presence of symptoms (syncope, aborted SCD, atrial fibrillation, or conduction defects), we refer to the syndrome. The question is if we can refer to having Brugada disease when a genetic cause for the syndrome is also found.
PhysiologyBrS has led to the discovery of new arrhythmia mechanisms: in particular, the phenomenon of phase-2 re-entry (P2R).5 The exact mechanism of VF in BrS is still debated. In addition to the classic re-entry based on abnormal conduction, P2R and the neural crest theory are 2 alternatives that may explain the arrhythmias. The Amsterdam group considers classic re-entry in the right ventricular outflow tract (RVOT) to be the main mechanism of VF, whereas the Utica group adhere to the P2R theory. Although, in the former mechanism, action potentials would be normal and the electric gradient would be caused by slow conduction of unsynchronized action potentials, in P2R the electric gradient is caused by a shortened duration of the action potential in the epicardium of the RVOT. In the first case, the problem lies in mutations that reduce sodium flux into the cardiac cell, in the second, it is caused by an excessive potassium flux (potassium ion gradient). Elizari's group proposes that the underlying cause of BrS is due to somatic mutations in neural crest cells. They consider BrS to be a developmental problem of the heart in the embryonic stage.6 This possibility of somatic mutations in BrS is also supported by the fact that approximately half of the patients are isolated cases and not familial, as if these patients were unable to transmit the disease via germinal cells. We should say, then, that BrS is a phenotype with many different possible causes.
GeneticsThe description of the first gene associated with BrS in 1998 marked an authentic historic landmark in the relationships between genetics and cardiology. Until then, genetic studies in cardiology were uncommon and the results were viewed more as a curiosity than as a potential contribution to understanding the mechanisms of the disease and developing a treatment. However, we then began to understand that, with certain mutations, the sodium channel remains open, repolarization is prolonged, and the patient develops long QT. If the sodium current decreases as a result of other mutations in the same gene, then conduction abnormalities and BrS develop. Suddenly, a completely new world opened up.
It is not surprising then that the number of published mutations for all hereditary cardiac disorders increased rapidly. With the new techniques for genetic testing (genome-wide association study), the whole diagnostic process has been greatly accelerated. All this new information comes with the issue of interpretation: do all mutations and all genes really matter? Are they the cause of the disease? What is the real relevance of polymorphisms? Unfortunately we do not have the resources, the time, or enough variety and number of patients to study the function of each mutation. There are models that can help us, but models always come with a certain degree of probability and uncertainty.
FertilityWith all its potential limitations, preimplantation genetic diagnosis (PGD) has become an obvious option for the treatment of hereditary diseases. Those who oppose the technique argue that hardly any diseases, especially BrS, are monogenetic. As well as the principal gene considered the cause of the disease, there must be other mutations and variations, including polymorphisms, which accumulate until reaching a certain level of genetic risk. Therefore, implanting an embryo selected based on the absence of a sodium channel mutation would have no value in preventing BrS. Using the same arguments, the defenders of PGD maintain that simply selecting an embryo without the mutation lowers this genetic risk, and as such reduces the risk of disease manifestation. PGD has been offered in our hospital for several years for more than 200 different diseases that are considered monogenetic, including BrS.7 If a class 4 or 5 mutation is identified, our usual practice is to offer PGD, in an attempt to avoid disease transmission.
PediatricsBrS is a cause of SCD in children and is also one of several possible causes of sudden infant death syndrome (SIDS). Few diseases have been the subject of such prolonged, esoteric speculation as SIDS. We now know that most of these sudden deaths are due to arrhythmias, including BrS.8
We face a similar problem in the diagnosis of epilepsy and syncope of unknown cause in children. Not only long QT syndrome, but also short QT syndrome and BrS should be included in the differential diagnosis, particularly in difficult-to-treat patients. We must also not forget that patients can have more than one disease concurrently: epilepsy and BrS,9 or vasovagal and arrhythmic syncope.
The diagnostic-therapeutic strategy for BrS in pediatric patients is especially delicate and there is a lack of scientific evidence. We face 2 issues: the strategy in children with a diagnosis of BrS, and the study of patients’ children. In the first instance, our approach is similar to that in adults, with close follow-up and implantation of an implantable cardioverter-defibrillator (ICD) if there are enough risk factors to make the patients high risk. In the case of patients’ children, we perform a pharmacological test at age 12 and 18 years or if there are symptoms. Up to 25% of the children with a negative test at age 12 years have a positive test at age 18 years, and symptoms can precede a positive test or the onset of a spontaneous type 1 pattern.10 We are particularly cautious in patients with sinus node dysfunction, as in our experience it is associated with a very poor prognosis and these children should receive an ICD.
Sports medicineNothing stirs the imagination more than the sudden death of a “perfectly healthy” athlete. Not all SCD in athletes occurs during exercise. In reality, the opposite is true.10 Most die suddenly after exertion, either immediately or later, at complete rest. It has long been known that long QT syndrome and catecholaminergic polymorphic ventricular tachycardia are causes. However, now, after a detailed study of the families of deceased patients, it appears that the most common cause is BrS.11
We consider it appropriate to perform at least 1 ECG in individuals who participate in regular sport. Hyperthermia during sport or vagotonic states after stopping exertion can induce a type 1 pattern or trigger syncope or SCD in patients with high-risk BrS. If there are symptoms, a family history of SCD at a young age, or an ECG with a type 2 pattern, we suggest performing a pharmacological provocation test.
Medicolegal medicineThe results of postmortems vary enormously from one study to another and with physicians’ experience and perseverance in finding a cause. Here, the study of relatives and the “molecular autopsy” come into play. The study by Papadakis et al.11 showed that the most common cause of SCD, when one could be found, was BrS. This was demonstrated with the results of the ajmaline test in relatives. Postmortem genetic tests can also reveal a possible causal mutation in 20% to 40% of cases.12
Preventive medicineInvestigating apparently healthy individuals is one of the best ways to discover occult diseases. However, the value of such detection can obviously depend on the investigator and the tests performed.
There is strong controversy in heart diseases: in favour of screening, the Italian studies13; against, the studies from the USA. Either way, there is a large difference in the arguments. While the Italian studies are based on a reduction in the incidence of SCD due to screening, the arguments from the USA against it are purely economic. But what price can we put on a young person's life?
Electronic cardiac devices and RVOT ablationBrS is a disease of young people. It can only be controlled (in terms of SCD) by implantation of an ICD. It is not surprising, then, that the implantation techniques have been adapted to children. For example, a subcostal abdominal implant is much more comfortable than a prepectoral implant, especially with regards to sports. The implantation can be done with epicardial leads, so that the patient's venous system remains completely intact. In experienced centers, this implantation can be combined with an RVOT epicardial ablation, where the substrate of BrS is located.14 This combination is our current protocol for the treatment of BrS. Of note, there are not yet sufficient data on the long-term effects of ablation. Currently, ablation is not an alternative to ICD.
Sex-related factorsIn our experience, the idea that BrS affects fewer women is false. Practically 50% of our patients are women.15 The fact that they show fewer symptoms could lead to fewer women being diagnosed, but with careful screening of relatives and a high clinical suspicion, the rate of diagnosis in women is similar to that in men.
Many publications on BrS have emphasized that male patients have a worse prognosis than female patients. While this appears to be true in adults, it is not the case before puberty. No differences have been found in symptoms or mortality between prepubescent boys and girls.10 It is very clear that testosterone has a role in BrS. Male castration has been demonstrated to improve the manifestations of the disease.16
Risk stratificationOne fundamental aspect of BrS is the assessment of risk of SCD. BrS has a very broad clinical presentation. The diagnosis may be made after an aborted SCD, but increasingly, patients are completely asymptomatic. Syncope, atrial fibrillation, sinus node dysfunction, and conduction abnormalities are symptoms and findings that affect the prognosis. But the question is, who should receive an ICD as prevention? Half of the patients with SCD had no previous symptoms.
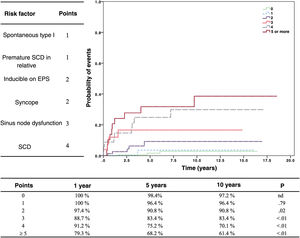
In an effort to integrate all the available information, we have developed a scoring system to determine the risk of arrhythmic events17 (figure 2). This risk stratification system is very valuable, but it also comes with a paradox: one may make the mistake of assuming that low risk means no risk. Patients with a low score are, therefore, not considered candidates for ICD protection, whereas patients with a high score automatically benefit from this protection. The result, paradoxically, is that patients with a high risk survive arrhythmic events due to the protection from their ICD, while those from the low-risk group, if they develop an arrhythmia, die due to lack of protection from an ICD. Thus, although the incidence of arrhythmias is much lower in the low-risk category, the true mortality is higher due to the lack of protection.

Risk stratification of patients with Brugada syndrome. On the left, the risk factors with their corresponding points. The graph shows the probability of arrhythmic events according to score. Reproduced with permission from Sieira et al.17 EPS, electrophysiological study; SCD, sudden cardiac death.
Over the past 30 years we have learned a lot about BrS, but also about other related diseases. It is clear that, with the description of BrS, the world of arrhythmology has entered a new dimension.
FUNDINGNo funding received.
AUTHORS’ CONTRIBUTIONSAll 3 authors contributed equally to the writing and review of the article.
CONFLICTS OF INTERESTP. Brugada has received compensation for teaching from Biotronik. C. de Asmundis receives research grants on behalf of his hospital from Biotronik, Medtronic, Abbott, Boston Scientific, AtriCure, Philips, and Acutus, and has received compensation for teaching and proctoring from Medtronic, Abbott, Biotronik, Boston Scientific, Atricure, Acutus Medical, and Daiichi Sankyo. J. Siera has no conflict of interests.