Keywords
INTRODUCTION
The syndrome of right bundle branch block, ST segment elevation and sudden cardiac death (SCD), better known today as Brugada syndrome, was described in 1992 as a new clinical and electrocardiographic syndrome involving susceptibility to ventricular arrhythmias and SCD in patients without obvious structural heart disease.1
The initial description, which included 8 patients, was followed by reports of new isolated cases,2,3 and numerous studies soon appeared mainly aimed at defining the clinical characteristics of larger series of patients4,5 or the genetic, cellular and molecular features of the disease.6-8 This review provides a summary of what is currently known about Brugada syndrome and up-to-date information from the main clinical and experimental studies published in recent years.
DEFINITION AND EPIDEMIOLOGY
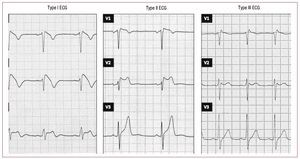
With more patients with Brugada syndrome being identified, certain questions soon arose regarding the definition of its characteristic electrocardiographic (ECG) pattern and the diagnostic criteria for the disease. Three different ECG patterns were described (Figure 1)9: a) type I, characterized by a coved-type ST-segment elevation ≥2 mm in more than one right precordial lead (V1-V3), followed by negative T waves; b) type II, characterized by ST-segment elevation ≥2 mm in right precordial leads followed by positive or biphasic T waves, resulting in a saddle-back configuration; and c) type III, defined as any of the 2 previous types if ST-segment elevation is ≤1 mm. Although the 3 patterns can be observed in Brugada syndrome, and even in the same patient at different times, type I is the only one that is considered diagnostic of the disease, as specified in the 2 consensus documents published in 2002 and 2005.9,10 Both documents stated that the definitive diagnosis of Brugada syndrome should only be established when the type I ECG pattern is documented in combination with at least one of the following clinical criteria: documented ventricular fibrillation (VF), documented polymorphic ventricular tachycardia (VT), inducible ventricular arrhythmias during electrophysiological study (EPS), syncope or nocturnal agonal respiration, a family history of SCD at <45 years of age, or type I ECG pattern in other family members.9,10 Nevertheless, this definition today is becoming outdated, particularly when we take into account that other important aspects of the disease are now known, such as causal mutations.11,12 In fact, our data demonstrate that a type I ECG pattern alone, even when other clinical criteria are not fulfilled, can be associated with SCD during follow-up.12 Thus, all patients who present a type I ECG pattern, even when isolated, should be considered at risk.

Figure 1. Electrocardiographic patterns (ECG) that can be found in the patients with Brugada syndrome. Type I only is diagnostic of the syndrome.
Brugada syndrome is included among the so-called channelopathies, that is, diseases produced by alterations in the transmembrane ion channels that participate in cell action potential, and which lead to an increased susceptibility to arrhythmias. Channelopathies are purely electrical disorders and are characteristically not associated with underlying structural heart disease. In fact, Brugada syndrome is the cause of 4% to 12% of all SCD and up to 20% of SCD that occur in normal heart.10
The prevalence of Brugada syndrome has been estimated at 5/10 000, although this figure possibly underestimates the actual prevalence, since many patients can present silent forms of the disease. Great geographical variability has been reported, such that the syndrome seems to be much more frequent in Asia than in western Europe or North America.13,14 In fact, the syndrome is thought to be endemic to certain regions of southeast Asia, where it is usually known as sudden unexplained death syndrome, also called bangungot (in Philippines), pokkuri (in Japan), or lai tai (in Thailand).15
GENETIC FACTORS UNDERLYING BRUGADA SYNDROME
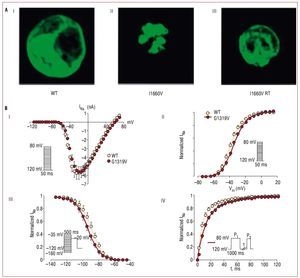
Brugada syndrome is typically transmitted via an autosomal dominant inheritance pattern.10 Nevertheless, the disease can be sporadic in a significant proportion of patients, that is, absent in other family members.16 The first mutations associated with Brugada syndrome were found in 1998 in the SCN5A gene (locus 3p21), that encodes for the cardiac sodium channel.6 To date, more than 100 different mutations leading to Brugada syndrome have been described in the same gene, whose effect, in all the cases studied, is a decrease in transmembrane sodium current (INa), either because of a quantitative reduction, or by a qualitative channel dysfunction (Figure 2).7,15,17-20 Even though SCN5 A is the only gene to have been associated with Brugada syndrome in almost a decade, only 18% to 30% of the patients test positive for mutations in this gene, which indicates that the disease is genetically heterogeneous.10 According to this hypothesis, 4 new genes associated with Brugada syndrome have been identified within the last 2 years, although how much they contribute to the total number of cases of the disease remains unknown. The first of them, GPD1-L (glycerol-3-phosphate dehydrogenase 1-like),21 was described by London et al in 2007, after having first identified the causal locus (3p22-p24) in 2002.22 The authors demonstrated that the mutation A280V in GPD1-L indirectly led to sodium channel loss of function, since it impaired the sodium channels trafficking to the cell membrane.21 Perhaps more novel is the finding of mutations in the CACNA1c and CACNB2b genes that encode the calcium channel23 and—quite recently—in the KCNE3 gene, that encodes for a b-subunit responsible for the transient outward potassium currents (Ito).24 Functional studies have demonstrated that in these cases, although the sodium channel is not affected, Brugada syndrome phenotype can be explained since a similar ion current imbalance occurs during phase 1 of the action potential.

Figure 2. Examples of 2 SCN5A gene mutations that decrease the sodium currents by different mechanisms. A: mutation I1660V produces a trafficking defect of the sodium channel to the cell surface; I, the wild-type channels (WT) are located both in the center and in the periphery; II, the channels with the mutation I1660V remain trapped in the intracellular organelles, and are not trafficked to the cellular membrane; and III, the mutated channels can be rescued by incubation at room temperature (modified by permission of Cordeiro et al18). B: mutation G1319V modifies the kinetic properties of the sodium channel; I, maximal peak current amplitudes are similar in mutated cells and in WT, indicating that the number of functional channels is similar in both cases; II, voltage-dependence of activation, showing a small depolarizing change in the mutated channels compared to WT; III, voltage-dependence of steady-state inactivation, reflecting enhanced inactivation in mutant channels; and IV, recovery after inactivation, that seems markedly slower in the mutated channels compared to WT (modified by permission of Casini et al19).
PATHOPHYSIOLOGY AND IONIC AND CELLULAR MECHANISMS
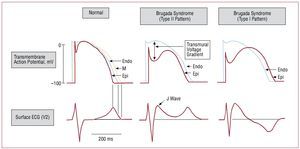
Various experimental studies have enabled the mechanisms involved in the development of the 2 main characteristics of Brugada syndrome to be elucidated, namely: the typical ECG morphology, and the susceptibility to VF and SCD. Figure 3 shows the normal action potential of ventricular cardiomyocytes and the ion currents involved in each of its phases. A decrease in INa, the disorder more frequently observed in mutations in SCN5A associated with Brugada syndrome,7,17-20 leads to an imbalance between the positive inward and outward currents at the end of phase 1 of the cell action potential. Figure 3 clearly shows that similar situations occur when there is a decrease of the inward L-type calcium current (ICa(L)) (produced by mutations in CACNA1c or CACNB2b)23 or an increase of the outward potassium currents (Ito) (produced by the mutation recently described in KCNE3).24 Whichever the mechanism, the imbalance between the inward and outward currents leads to the development of a characteristic notch and the loss of the action potential dome mediated by an increase (relative or absolute) of the outward Ito currents. Since the density of Ito is greater in epicardium than in endocardium, this event occurs heterogeneously on the ventricular wall and leads to a transmural voltage gradient, which produces the characteristic ST-segment elevation in the ECG (Figure 4).8

Figure 3.Transmembrane action potential and ion currents that participate in each of its phases. The shaded area corresponds to phase 1, mainly determined by the balance between the positive inward INa and ICa(L) currents and the positive outward Ito currents. When the outward currents predominate over inward currents (*), the cell undergoes a certain degree of repolarization, which produces the characteristic notch in the action potential (discontinuous line).

Figure 4.Proposed mechanism of ST-segment elevation in Brugada syndrome. The appearance of a notch in certain regions of the epicardium, but not in the endocardium, creates a transmural voltage gradient, which produces J-point elevation. If the notch is marked, the action potential in the epicardium is lengthened compared to the endocardium, which causes ST-segment elevation and appearance of negative T-waves. Modified by permission of Antzelevitch.25 Endo indicates endocardium; Epi, epicardium; M, myocardium.
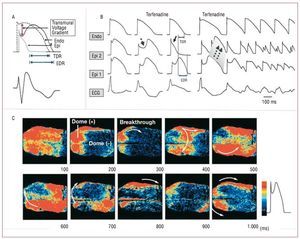
The ion current imbalance at the end of phase 1 of the action potential also explains the susceptibility to develop ventricular arrhythmias in Brugada syndrome, which would arise via a phase 2 reentry mechanism. In circumstances where the notch reaches approximately -30 mV, all-or-none repolarization occurs, which can lead to the complete loss of the action potential dome.25 This event also takes place heterogeneously between the epicardium and endocardium and even between various points in the epicardium, leading to transmural and epicardial dispersion of repolarization, respectively (Figure 5). This creates a favorable substrate for the onset of premature ventricular complexes as a consequence of the propagation of the action potential dome from sites at which it is maintained to sites where it has been lost (Figure 5B).8,25 This hypothesis has been confirmed by high-resolution optical mapping studies conducted in canine right ventricular samples, which found a gradient between the regions with and without a dome in the action potential and the development of a reentrant circuit initially limited to the epicardium that gradually involved the endocardium (Figure 5C).26

Figura 5. Proposed mechanism of ventricular arrhythmias in Brugada syndrome. A: a further shift in the balance of inward and outward currents at the end of phase 1 produces all-or-none repolarization; in these circumstances the action potential dome can disappear completely (gray line), which causes transmural (TDR) and epicardial (EDR) dispersion of repolarization (modified by permission of Antzelevitch25). B: action potentials recorded in endocardium and in 2 epicardial sites in canine right ventricular samples; the administration of terfenadine, a sodium and calcium channel blocker, accentuates the notch in the epicardial action potential and produces an all-or-none repolarization; this situation facilitates propagation of the action potential dome from the regions where it is maintained to the regions that have lost it, which leads to a premature ventricular beat produced by a phase 2 reentry mechanism (dashed arrows), that can trigger a polymorphic ventricular arrhythmia (modified by permission of Antzelevitch25). C: high-resolution optical mapping with simultaneous registry of 256 action potentials in canine right ventricular samples; in line with the explanation offered in B, propagation can be observed from regions indicated in red (that maintain the action potential dome) toward those in blue (without the action potential dome) (modified by permission of Shimizu et al26).
The concept of an imbalance between inward and outward ion currents, defining the pathological substrate of Brugada syndrome, has many applications. Firstly, it assists in the development of experimental models of the disease, which can be created via the administration of drugs that open the outward potassium currents,8 sodium channel blockers8 or combined sodium and calcium channel blockers,27 among others. Furthermore, it explains the effect of specific modulators and certain special features of the syndrome, such as increased phenotypic expression (and the risk of arrhythmic complications) during vagal activity28-30 (acetylcholine blocks the calcium currents, whereas betamimetics increase them),31 or the greater severity of the syndrome among men than in women32 (men may have a constitutionally higher density of Ito than women).33 On the other hand, maneuvers that increase ion imbalance are not recommended for patients with Brugada syndrome, such as the administration of sodium channel blockers, although at the same time these drugs may be useful in unmasking weakly expressed forms of the syndrome (see section: Diagnostic Tools: Drug Challenge Test).34 In contrast, drugs with the opposite effect, therefore ones that restore ion balance, could have an application in the treatment of patients with Brugada syndrome. In this regard, the first results have been obtained using Ito blockers (such as quinidine)35 or ICa(L) activators (such as isoproterenol)36 (see section: Treatment).
CLINICAL ChARACTERISTICS
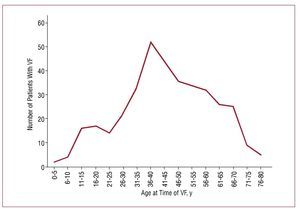
Patients with Brugada syndrome usually remain asymptomatic. Nevertheless, it has been reported that 17% to 42% of them present syncope or SCD as a consequence of a ventricular arrhythmia at some time during their lives.37-40 This figure probably overestimates the real incidence of events, given that many asymptomatic patients are not diagnosed. Patients usually present symptoms, especially SCD, during their fourth decade,16 although no conclusive explanation for this has been offered to date (Figure 6). Approximately 23% of the patients with SCD had already undergone syncope.38

Figure 6. Incidence of sudden death (SCD) or documented ventricular fibrillation (VF) according to age at presentation. Updated data of 370 patients taken from the international registry. Cardiac events occurred in a total of 120 patients (32.4%).
Given that a significant number of patients, around 20%, may suffer supraventricular arrhythmias, mainly atrial fibrillation (AF),41 some patients can experience palpitations and dizziness. Other symptoms, such as neurally mediated syncope, have also been reported in isolated cases of Brugada syndrome.42,43
As occurs in other channelopathies with sodium channel disorder, arrhythmias in Brugada syndrome (and, thus, the symptoms) typically appear during predominant vagal activity, such as rest, or even during sleep.28 In a study by Matsuo et al,29 26 of the 30 episodes of VF documented by automatic implantable cardioverter-defibrillator (ICD) occurred at night, which has been confirmed in more recent series.30 As mentioned, increased vagal tone mediated by acetylcholine decreases the calcium currents, which could lead to arrhythmogenesis via phase 2 reentry.31 On the other hand, a recent work using positron emission tomography demonstrated that the patients with Brugada syndrome present a certain degree of sympathetic dysfunction, which manifests as a decrease in norepinephrine at the synaptic cleft, which also favors arrhythmogenesis through a decrease in intracellular cyclic adenosine monophosphate (AMP).44
The Brugada syndrome phenotype is thought to be 8 to 10 times more prevalent in men than in women.10 As proof of this, approximately 71% to 77% of patients diagnosed with Brugada syndrome are men, a finding that is regularly observed in all series.37-40 In a recent study that included a series of patients who were followed up for the longest period to date, our group observed that, at the time of diagnosis, men more frequently presented previous symptoms and a spontaneous type I ECG pattern and, in addition, developed greater inducibility of VF during the EPS as compared to women (Figure 7A).32 During follow-up, behavior also differed according to sex. Of the 272 men included in the study, 31 (11.6%) had documented SCD or VF during an average follow-up period of 58 (48) months, whereas the rate of events in the female population during the same follow-up period was considerably lower (3/112 [2.8%]; log-rank test P=.007) (Figure 7B).32

Figure 7.Differences in Brugada syndrome patterns between men (M) and women (W). A: clinical characteristics at the time of the first clinical assessment. B: survival analysis of major cardiac events defined as sudden cardiac death (SCD) or ventricular fibrillation (VF) during follow-up. In total, 31/272 men (11.6%) and 3/212 women (2.8%) presented major events during an average follow-up of 58 months. Data obtained from Benito et al.32
Two different hypotheses have been proposed to explain why Brugada syndrome is more strongly expressed in men than in women. On the one hand, it has been demonstrated that there are constitutional differences in the transmembrane ion currents between sexes. In a study conducted with canine samples, Di Diego et al33 verified that the density of epicardial Ito currents is significantly higher in men than in women,33 which, according to the theory of ion imbalance in phase 1, predisposes to greater ST-segment elevation and greater susceptibility to the onset of ventricular arrhythmias (see section "Ion and Cellular Pathophysiology and Mechanisms"). On the other hand, there are suggestions that hormonal effects may play a role in the phenotypical differences between sexes. In this regard, disappearance of the ECG type I pattern has been reported after castration in patients with Brugada syndrome and concomitant prostate cancer45 and, on the other hand, testosterone concentrations seem to be significantly greater in men with Brugada syndrome than in controls.46 Some experimental studies suggest that hormones could exert their effect by modifying ion currents.47,48 In line with the hormonal hypothesis, the current data, although sparse, on the pediatric population with Brugada syndrome do not indicate differences in behavior between boys and girls before 16 years of age.49
In fact, although 3 of the 8 cases initially published in the first description of the syndrome were children, to date there has been little information on the behavior of Brugada syndrome in the pediatric population. Probst et al49 recently presented the results of 30 children less than 16 years of age included in a multicenter study. More than half of the patients (n=17) had been diagnosed during family screening, although 11 patients had already experienced symptoms. They point out that, of the 11 symptomatic patients, 10 had a spontaneous type I ECG pattern and, characteristically, in 5 of them the symptoms had appeared in association with febrile episodes. An ICD was implanted in 5 patients and treatment with hydroquinidine was begun in 4. During a mean follow-up of 37 (12) months, 3 patients (10% of the population) underwent SCD (n=1) or received an appropriate ICD shock (n=2).49 The 3 patients had previous syncope and all had a spontaneous type I ECG pattern. It should be pointed out that the 4 patients treated with hydroquinidine remained asymptomatic throughout follow-up.49 We obtained similar results in population of 58 patients under 18 years of age.50 In our series, 6 patients presented major cardiac events (2 SCD and 4 VF) during a mean follow-up of 48.8 (48) months. Although cardiac events appeared more often in the patients with a spontaneous ECG type I pattern or inducibility during the EPS, the presence of previous symptoms was the variable most associated with prognosis in our pediatric series (Figure 8).50 These 2 studies, although small, indicate the following: a) Brugada syndrome may manifest in the pediatric age group; b) febrile episodes are a frequent trigger of arrhythmias in children with Brugada syndrome; c) symptomatic patients, especially if presenting a spontaneous ECG type I pattern, constitute a group with particularly high risk of ventricular arrhythmias within a relatively short follow-up period; and d) the patients with worse prognosis benefit from ICD implantation, although quinidine treatment may be proposed as an alternative, especially in the youngest patients.

Figure 8. Survival analysis of major cardiac events in the pediatric population with Brugada syndrome. Data obtained from 58 patients.50 The greater event rate was observed among the symptomatic patients, who in turn more frequently presented a spontaneous Type I electrocardiographic pattern and inducibility of arrhythmias in the electrophysiological study.
ELECTROCARDIOGRAM AND MODULATING FACTORS
As mentioned, the only pattern which is definitive and diagnostic of Brugada syndrome is a type I ECG pattern (Figure 1). However, particular clinical situations can lead to a similar ECG pattern (Table 1). In some cases, this is due to completely independent conditions which lead to ECG findings resembling those of Brugada syndrome (thus, these are conditions that should be ruled out in the differential diagnosis), whereas in other cases ST-segment elevation is particularly evident when there is a genetic predisposition.16

It is relevant to emphasize that the ECG of patients with Brugada syndrome can vary over time and, thus, can show type I, II, and III patterns in a single patient at different times or even be temporarily normal.51 Thus, serial electrocardiograms are recommended in all patients.51,52 There are numerous modulating factors that to a certain extent may explain the variability of the electrocardiogram. The factors listed in the second column of Table 1 can increase ST-segment elevation in the patients with known Brugada syndrome, since they exacerbate ion current imbalance during phase 1 of the myocardial action potential.16,25,34 For the same reason, autonomic tone and the influence of certain hormones can likewise modulate ST-segment elevation and may even explain the greater rate of occurrence of arrhythmias in particular conditions (see section: Pathophysiology and Ion and Cellular Mechanisms).28-30,45,46 Temperature can be an important modulating factor in some patients with Brugada syndrome. It has been demonstrated that, in some SCN5A mutations, a temperature increase accentuates the premature inactivation of the sodium channel.53 This explains why in some patients febrile episodes can unmask silent forms of Brugada syndrome and confer an increased risk (transient) of ventricular arrhythmias,54,55 which seems to be especially important in the case of the pediatric population.49
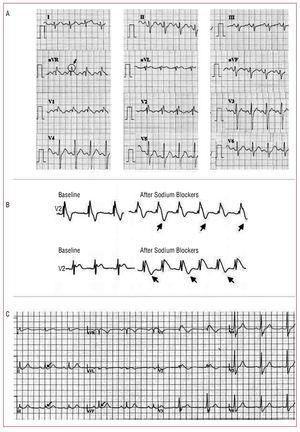
In recent years, numerous studies have attempted to identify new characteristic ECG features and their possible prognostic significance. Pitzalis et al56 described a corrected QT interval (QTc) prolongation in right precordial leads (but not in left precordial leads) in patients with Brugada syndrome, particularly after the administration of sodium channel blockers. Subsequently, this was correlated with a worse prognosis, especially if the duration of the QTc interval in V2 is ≥460 ms.57 Similarly, the aVR sign (defined as the presence of an R wave ≥3 mm or a R/q ratio ≥0.75 in lead aVR) has been associated with an increased risk of ventricular arrhythmias (Figure 9A).58 In these cases, it is thought that the increase in the R wave probably indicates greater ventricular conduction delay and, thus, greater electrical heterogeneity.58 The presence of T-wave alternans, also a sign of transmural dispersion of repolarization,59 can be observed in patients with Brugada syndrome after the administration of sodium channel blockers and, in addition, makes it possible to identify a subgroup at greater risk of VF during follow-up (Figure 9B).60 On the other hand, and very recently, it has been reported that up to 11% of the patients with Brugada syndrome can present an early repolarization pattern in inferior or lateral leads, which is also associated with a greater rate of symptoms (Figure 9C).61

Figure 9. Incidental electrocardiographic findings that have been associated with a greater risk of arrhythmias in patients with Brugada syndrome. A: aVR sign (reproduced by permission of Babai et al58). B: T-wave alternans, that can appear after administration of sodium channel blockers (modified by permission of Tada et al60). C: early repolarization pattern in inferior and lateral leads (reproduced by permission of Sarkozy et al61).
Conduction disorders can sometimes be observed in the ECG of patients with Brugada syndrome. In fact, the decrease in sodium currents can lead to both phenotypes (Brugada syndrome and conduction disorder), either in isolation or within the same family.62 Thus, it has been reported that parameters such as the PQ interval, the duration of the QRS complex or the HV interval are more prolonged in patients who have an identified mutation in the SCN5A gene (and, thus, sodium channel disorder) than in patients who test negative for an SCN5A mutation.63 In a recent study with 200 patients with Brugada syndrome, our group verified that certain conduction disorders, such as prolonged QRS complex, are observed more often among symptomatic patients than among asymptomatic ones. In this population, a QRS cut-off point of ≥120 ms effectively predicted an odds ratio (OR) = 2.5 (95% confidence interval [CI], 1.4-4.6; P=.003) of being symptomatic.64 It seems that women with Brugada syndrome are more susceptible to conduction disorders than men.32 In fact, the administration of sodium channel blockers during a challenge test in women leads to a significantly greater increase in the PR interval and QRS duration.32 This is in agreement with the results of previous experimental studies demonstrating that ventricular samples obtained from canine females expressed a lower Ito current those obtained from males, which explained the
predisposition to conduction disorders in the former and, on the other hand, greater ST-segment elevation in the latter.65
DIAGNOSTIC TOOLS: DRUG CHALLENGE TEST
Given that the ECG pattern of patients with Brugada syndrome varies over time and even can be temporarily normal, the use of drug challenge tests has grown in recent years. Sodium channel blockers are the most often used drugs, mainly because they are effective, easily available, and have rapid activity.34 The administration regimens of the main sodium blockers used as a diagnostic test for Brugada syndrome are outlined in Table 2.10 Brugada syndrome is confirmed if, after testing with any of these drugs, an ECG pattern defined as type I (Figure 1) appears or is accentuated. The test should be performed using continuous monitoring, taking, an electrocardiogram every minute until the end. It should be terminated when: a) type I ECG pattern appears, thus confirming the diagnosis; b) multiple extrasystoles or other ventricular arrhythmias appear; or c) QRS undergoes widening >130% compared to baseline.10

Current data indicate that ajmaline is the most effective drug in the diagnosis of Brugada syndrome. In a study with 147 individuals with an identified SCN5A gene mutation, the drug challenge test using ajmaline had a sensitivity, specificity, positive predictive value and negative predictive value of 80%, 94.4%, 93.3%, and 82.9%, respectively, in the diagnosis of Brugada syndrome.66 These figures are considerably higher than those obtained using flecainide in another study with 110 genotyped patients, whose sensitivity, specificity, positive predictive value and negative predictive value were of 77%, 80%, 96%, and 36%, respectively.67 The low negative predictive value found in this study is noteworthy, and should be taken into account whenever a flecainide test is performed, especially in particular contexts, such as in family screening.16 The diagnostic values of ajmaline and flecainide were directly compared in a recent study, in which 22 patients with confirmed Brugada syndrome underwent successive challenge tests using the 2 drugs. Whereas the test confirmed the diagnosis in all 22 patients when ajmaline was used, only 15 patients showed a positive test after flecainide administration.68 Furthermore, ST-segment elevation using ajmaline (0.43 [0.15] mV) was greater than that obtained using flecainide (0.29 [0.18] mV). Patch-clamp studies have verified that flecainide, in addition to blocking the sodium channel, decreases the Ito currents to a greater extent, which explains its reduced effectiveness compared to ajmaline.68
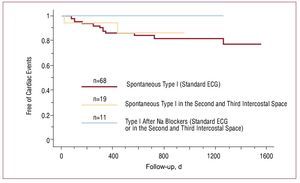
Due to the limited value of the standard electrocardiogram, even when facilitated by drug challenge, new strategies have been proposed to help in the diagnosis of Brugada syndrome. It has been demonstrated that positioning the right precordial leads in higher intercostal spaces (third and even second intercostal space) increases sensitivity in relation to the baseline electrocardiogram and after administration of sodium channel blockers.69 Recent data demonstrate that a type I ECG pattern obtained in the second and third intercostal spaces, even when the electrocardiogram carried out with standard leads is normal, helps to identify a subgroup of patients whose prognosis is comparable to that of patients with a type I ECG pattern in the standard leads (Figure 10).70 Thus, this strategy facilitates identifying patients at risk who would not have been otherwise identified.

Figure 10.Survival analysis of cardiac events (sudden death or documented ventricular fibrillation) in patients with a spontaneous type I electrocardiographic pattern obtained in standard leads, in patients with spontaneous type I electrocardiographic pattern which only appears by raising the right precordial leads to the second or third intercostal space, and in patients who develop a type I pattern after administering sodium channel blockers. No differences in prognosis were found between the first 2 groups (modified by permission of Miyamoto et al70)
PROGNOSIS AND RISK STRATIFICATION
Due to the great phenotypic variability of patients with Brugada syndrome, that ranges from the absence of symptoms to SCD at an early age, the search for parameters to help stratify risk has been of great interest in recent years.37-40 However, the published studies have not obtained similar results, and currently risk stratification in patients with Brugada syndrome continues to be a matter of debate in certain aspects.
In the last series presented by Brugada et al71 on the international registry database, the percentage of individuals who had documented SCD or VF at some time during their life was 25% (178 of 724 patients). Undoubtedly, this figure overestimates the real incidence of events, because, on the one hand, this series included the first patients identified in the first years after the disease was described (usually the period when only the clearest cases are identified) and, on the other, because the population in the international registry is usually at greater risk.16 However, in this series, it is important to note that the incidence of major events ranged between 3% and 45%, according to the baseline characteristics of the patients. This justifies performing risk stratification in all patients with Brugada syndrome.
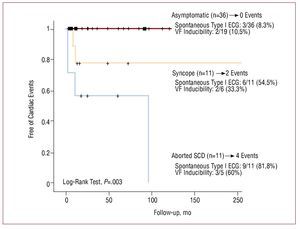
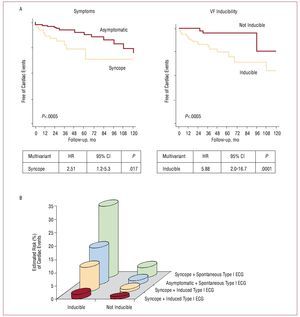
Aborted SCD is an indisputable risk factor and recognized by all studies.37-40 Data from Brugada et al37 confirm that, among patients who have undergone aborted SCD, 62% present a new arrhythmia during a mean period of 54 months. This means that these patients should be protected with an ICD as secondary prevention (class I indication).10 In patients without previous cardiac arrest, Brugada et al reported that the presence of previous syncope, a spontaneous type I ECG pattern and the inducibility of ventricular arrhythmias in the EPS are also prognostic markers. In a population of 547 patients (mean age, 41 [45] years; 408 men, 423 asymptomatic and 124 with previous syncope; 71.5% with spontaneous type I electrocardiogram at baseline), 45 individuals (8.2%) presented a first major cardiac event (documented SCD or VF) during a mean follow-up of 24 (32) months.39 Univariate analysis associated a history of previous syncope (hazard ratio [HR] = 2.79 [95% CI, 1.5-5.1]; P=.002), the presence of a spontaneous type I ECG pattern (HR=7.69 [95% CI, 1.9-33.3]; P=.0001), male sex (HR=5.26 [95% CI, 1.6-16.6]; P=.001) and the inducibility of ventricular arrhythmias in the EPS (HR=8.33 [95% CI, 2.8-25]; P=.0001) with the appearance of events during follow-up.39 Multivariate analysis confirmed the presence of previous syncope and the inducibility of VF in the EPS (Figure 11A) as independent predictors of prognosis. Logistic regression analysis assisted in defining 8 risk categories according to the presence of symptoms, the findings of the baseline electrocardiogram, and the results of the EPS (Figure 11B). Subsequent data analysis identified the EPS as particularly useful in the stratification of asymptomatic patients and without a family history of SCD (known as fortuitous cases, n=167). In fact, 11 of 167 patient (6%) presented VF during follow-up and inducibility in the EPS was the only factor associated with prognosis in this subgroup of patients.71

Figure 11. Major cardiac events (sudden death or documented ventricular fibrillation) during follow-up of patients without previous cardiac arrest. A: event survival analysis according to the presence of previous symptoms and inducibility of ventricular arrhythmias in the electrophysiological study. B: probability of events during follow-up estimated by logistic regression analysis, according to the presence of symptoms, inducibility of arrhythmias and type of baseline ECG (data obtained data from Brugada et al39).
Other groups agree that previous symptoms and a spontaneous type I ECG pattern are risk markers in patients with Brugada syndrome, although, in general, they have reported a lower general incidence of events (6.5% in the series of Priori et al,38 with a mean follow-up of 34 [44] months, and 4.2% in the series of Eckardt et al,40 with a follow-up of 40 [50] months). This is probably due to the fact that the series of Brugada et al included populations of patients with more severe disease.40 The other groups also agree that the inducibility of arrhythmias in the EPS is greater among patients with syncope or MS38,40 but, in contrast to the series of Brugada et al, have not demonstrated that the EPS has a role as a prognostic tool. There may be several reasons to explain this difference:71 a) the use of nonstandard ventricular stimulation protocols due to including patients from several centers; b) the inclusion, in some series, of patients with a type II or type III ECG pattern, and thus, without definite confirmation of the syndrome; and c) the absence of events during follow-up in the other series. The last point may vary when longer follow-ups are available, since events (and along with them the positive predictive value) can only increase with time.71
Since the first series were described, it has been noted that Brugada syndrome appears in a more aggressive form among men than among women. Our group recently analyzed this observation in 384 patients (272 men and 112 women), recruited from just 2 reference centers (Hospital Clínic de Barcelona, Barcelona, Spain and UZ Brussels, Belgium) to avoid the selection bias that has been attributed to the international database, which mainly consists of high-risk patients.32 As mentioned, (see section: Clinical Characteristics), men and women presented different baseline characteristics and a strikingly different behavior during follow-up (Figure 7). This study also contributed the new information that risk markers could also be different between sexes.32 In fact, the factors associated with poor prognosis described for mixed populations (symptoms, spontaneous type I ECG pattern and inducibility in the EPS) were confirmed as valid for stratifying risk in men.32 In contrast, and taking into account the extremely low event rate in the women, none of these variables had sufficient power to identify those at greater risk. In the female population, however, conduction disorders seemed to be more associated with the event rate, and the PR interval specifically was the only independent predictor of risk in women.32
The presence of AF, which may be spontaneously found in 10% to 54% of the patients with Brugada syndrome,41,72 has recently been associated with worse prognosis. In a series of 73 patients, Kusano et al72 found that the patients with documented spontaneous AF present a greater incidence of syncope (60%) and documented VF (40%) than the patients with no evidence of AF (22% and 14%, respectively; P<.05). Our data indicate that this is the case in the male population as well as in the female population.32 As mentioned, other ECG findings may have some prognostic significance, among which are prolonged QTc interval in V2, the aVR sign, the presence of T-wave alternans, a repolarization pattern in inferior or lateral leads, and the width of the QRS complex (see section "Electrocardiogram and Modulating Factors").57-61,64
Neither a family background of SCD nor the presence of SCN5A gene mutation have been defined as risk factors in any of the large series reported to date. However, recent studies indicate that other genetic findings could well have prognostic significance. In a study that included 147 patients with Brugada syndrome or progressive conduction system disease, together carrying 32 different SCN5A gene mutations, Meregalli et al73 reported that the patients carrying a mutation resulting in premature stop codon (whose final effect is the production of a truncated protein) presented greater rates of syncope than the patients carrying any other type of mutation (25.3% vs 5.7%, respectively; P=.03). However, the authors could not demonstrate differences in the rate of greater arrhythmic complications (SCD or VF) according to the type of mutation. Our data on 188 patients (all with Brugada syndrome), together carrying 69 different SCN5A gene mutations, did in fact demonstrate differences in the major events rate, defined as SCD or VF at some time during their lifetime (truncated mutation vs other mutations, 23.9% vs 7.7%; P=.01).74 On the other hand, it seems that the presence of certain polymorphisms could modulate risk among patients with Brugada syndrome. In this regard, we have observed that the concomitant presence of the H558R polymorphism and a SCN5A gene mutation is associated with a more benign phenotype.75 In general, the search for genetic parameters with prognostic value is especially attractive, given that genetic information, in contrast to clinical information, is constitutional and, thus, invariant in the same individual.
TREATMENT
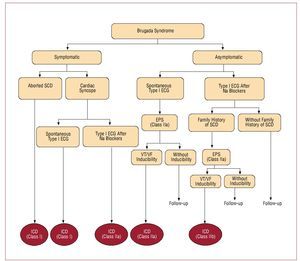
An ICD is the only treatment with demonstrated efficacy in Brugada syndrome. The current indications for ICD follow the recommendations of the II International Consensus published in 200510 (Figure 12). In general, ICD implantation is recommended for all patients who have already had symptoms and for asymptomatic patients in whom the EPS induces ventricular arrhythmias, especially if they present a spontaneous type I ECG pattern. In the asymptomatic patients, without a family history of SCD and whose type I ECG pattern is only documented after the administration of sodium channel blockers, periodic follow-up is recommended without the need of an EPS for risk stratification.10

Figure 12. Indications for automatic implantable cardioverter-defibrillator (ICD) in patients with Brugada syndrome (adapted by permission of Antzalevitch et al10).
Taken together, the 2 main retrospective studies conducted with patients with Brugada syndrome implanted with an ICD show that the rate of appropriate shocks is 3.7% per year.76,77 It should be noted that this rate is not only comparable to that described in other studies conducted on other heart diseases,78,79 but in this case it also refers to a young, and, furthermore, healthy population whose life expectancy could be higher than 30 years. However, and possibly due to the fact that a young active population was involved, the rate of inappropriate shocks was considerable (20% in the study of Sacher et al76 and 36% in the study of Sarkozy et al77). The leading causes of inappropriate therapy were, in both studies, sinus tachycardia, supraventricular arrhythmias, T-wave oversensing and lead failure.76,77 Thus, and because the ICD is not universally applicable therapy, in recent years a special effort has been dedicated to searching for possible drug options for the treatment of Brugada syndrome.
With the aim of reducing the ion imbalance at the end of phase I of the action potential, 2 main strategies have been proposed: a) the use of potassium current (Ito) blockers, and b) the use of drugs that increase the calcium (ICa(L)) currents. It has been demonstrated that quinidine, an antiarrhythmic agent that blocks Ito currents, reduces the incidence of induced arrhythmias in patients with Brugada syndrome,35 and it has been used successfully in specific clinical situations, such as the treatment of arrhythmia storms.80 Furthermore, its usefulness has recently been demonstrated as adjunctive therapy to ICD in patients with multiple shocks81 or as a therapeutic alternative to ICD in children at risk of arrhythmias.49 In turn, betamimetic drugs, such as isoproterenol, that increase inward ICa(L) currents, have been used with good results in cases of arrhythmia storm.36 Finally, the use of cilostazol, a phosphodiesterase III inhibitor that decreases Ito and increases ICa(L), has recently emerged as a promising therapy, although the few clinical cases published to date report inconsistent results.16
Correspondence: Dr B. Benito,
Electrophysiology Research Program, Research Center, Montreal Heart Institute,
5000 Rue Belanger, Montreal (QC), H1T 1C8 Canada
E-mail: bbenito@clinic.ub.es