Keywords
INTRODUCTION
Macroscopic findings define cardiac hypertrophy (CH) as a thickening of the interventricular wall and/or septum1; in the cell, it is characterized by an increment in cardiomyocyte size, with increased protein synthesis and changes in the organization of the sarcomeric structure.2 Although CH initially constitutes a compensatory response that transiently normalizes the biomechanical stress and optimizes cardiac pump function, prolonged hypertrophy is a highly important risk factor for the development of heart failure.3,4 Despite the fact that the dichotomy between adaptive and nonadaptive hypertrophy was described more than a century ago (Osler, 1892), the mechanisms that cause the progression of hypertrophy to heart failure have yet to be clearly understood.
From a phenotypic point of view, there are 2 types of CH, one concentric, secondary to pressure overload and characterized, within the cell, by the addition of sarcomeres in parallel, with the cardiomyocytes growing in the lateral direction, and the other, excentric, owing to volume overload, characterized by the addition of sarcomeres in series, with longitudinal cell growth.5 In molecules, these changes in cell phenotype are accompanied by a reinduction of the so-called "fetal program," the expression pattern of which, as we will show later, is in some aspects similar to that observed during embryonic development.2
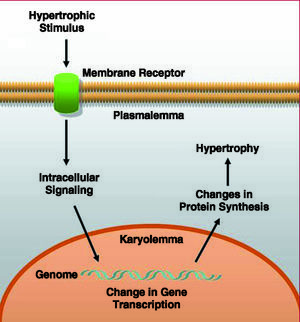
Cardiac hypertrophy is present in a number of heart diseases, including ischemic heart disease, hypertension, heart failure and valve diseases. From the pathogenic point of view, these different disease processes induce cardiomyocyte growth, due either to an increase in mechanical stress or in response to an increase in neurohormonal stimulation. This distinction, however, is a question of didactics since, under pathophysiological conditions, both types of stimuli are present simultaneously, and the intracellular pathways that are activated share a number of interconnections.6-8 These processes ultimately lead to the activation of the immediate early genes (c-jun, c-fos, c-myc) and the fetal genes (atrial natriuretic factor [ANF], beta-myosin heavy chain [β-MHC], and skeletal alpha actin [SKA], among others), which are considered and employed as hypertrophic markers8 (Figure 1).

Figure 1. General diagram of the events taking place in the process of cardiomyocyte hypertrophy.
Although the concept of the heart as a postmitotic organ has recently been questioned,9-11 it is widely accepted that the cardiomyocyte does not reenter the cell cycle. Thus, many of the intracellular signaling pathways that regulate cell proliferation in other cell types, in the cardiac myocyte, modulate hypertrophic growth.12,13 This effect occurs through the convergence of cytoplasmic signaling events and cascades in the nucleus where, among other things, they activate or inhibit proteins capable of regulating gene expression known as transcription factors. There is a considerable variety of transcription factors that appear to play a role in the regulation of the events that lead to the activation of the genetic program involved in CH.14-16 In recent years, it has become increasingly evident that the different transcription factors participating in the hypertrophic response do not act independently, but share a number of structural and/or functional interactions. In cardiac muscle, the true magnitude of these events is beginning to be understood, and they appear to be a lot more complex than those that take place in other cell types, such as skeletal myocytes.17,18
The traditional notion that CH has a compensatory role has recently been questioned,2 a circumstance that has stimulated research focusing on the molecular events implicated in their nonadaptive aspects, such as arrhythmogenicity and the development of heart failure. To advance our understanding of this condition, it is also necessary to identify in detail the similarities and differences between the signaling systems responsible for promoting the generation of a pathological, versus a physiological, hypertrophic response. Together, these new research lines may enable us to modulate hypertrophic growth, achieving a clinical benefit without provoking hemodynamic compromise.
We provide a synoptic view of the molecular events that take place in the pathological hypertrophic response, with special emphasis on the new lines and directions being introduced in basic research in recent years.
THE HYPERTROPHIC STIMULUS AND ITS RECEPTORS
Biomechanical stress can be produced in the myocardium by means of 2 types of noxious events: mechanical stretch and the release of neurohormonal factors.7 The mechanical stress induced by the physical stretching of adult and neonatal cardiomyocytes is sufficient to induce a phenotypic and genetic hypertrophic response, even in the absence of humoral and neuronal factors.8 Despite its pathophysiological importance, little is known about how this biomechanical stress is perceived by the cardiomyocyte and how this is translated into prohypertrophic intracellular signals. Although the mechanical stimulus activates multiple messengers and intracellular signaling pathways, we do not know for certain which are activated directly and which are stimulated through other agents. Nor is the time sequence of the events clear since the initial moment of second messenger activation has not been determined systematically.
It is traditionally assumed that, in order for a mechanosensitive molecule to quantify plasma membrane stress, there should be some type of interaction between the two. Stress has long been recognized as a biochemical regulator of the activity of many types of cells such as, for example, the skeletal myocyte,19 in which the force generated on the membrane is sufficient to alter a number of enzyme activities.20 As a result, several types of molecules have been proposed as candidates to fulfill this role, in particular the ion channels,21,22 integrins,23 and tyrosine kinases.24 The sarcomeric Z disc has been the object of increasing interest as a candidate to carry out the task of mechanotransduction for several years now.25,26 Mouse cardiomyocytes lacking the MLP protein (muscle LIM protein), which is normally present in the Z disc, exhibit a selective absence of the response to mechanical stretch, but respond normally to G protein-coupled receptor agonists; on the other hand, in humans, a MLP gene mutation results in telethonin/ T-cap bond breakage, which leads to dilated cardiomyopathy.26 Frey et al27 recently reported a novel family of proteins specific for sarcomeric Z-disc of striated muscle, referred to as calsarcins, which are capable of interacting with both telethonin/T-cap and calcineurin, a circumstance that indicates a possible role as a link between the capture of the mechanical stimulus and hypertrophic signaling.27
Despite our limited knowledge concerning its receptors and transduction pathways, the importance of the mechanical stimulus in triggering the hypertrophic process is supported by a large body of evidence from studies of different types.8 In addition, it is now accepted that mechanical stress acts as the initial stimulus in the series of events that results in in vivo myocyte growth.6 This has led to the search for better experimental models that enable us to faithfully mimic the in vivo situation of the cardiomyocyte, resulting in the availability of a considerable number of methodological approaches for the mechanical stimulation of these cells.
The changes in the myocardium of a hypertensive patient occur not only in left ventricle, which is directly exposed to the hemodynamic overload, but in the interventricular septum and right ventricle, as well.7 On the other hand, the infarcted myocardium presents phenotypic alterations in regions far from the necrosed tissue. This evidence indicates that, together with the mechanical component that acts in the myocardial region that is directly subjected to stress, humoral factors intervene, exerting their action in a more diffuse manner.7 There are a number of humoral factors that can act as a hypertrophic stimulus in the cardiomyocyte, and the consequences of their activation differ. A first group consists of the growth factors (transforming growth factor-beta [TGF-ßβ, fibroblast growth factor [FGF], and insulin-like growth factor [IGF], among others)28,29 which, through their action in membrane receptors with tyrosine kinase activity, activate an intracellular second messenger cascade which ultimately leads to a normal growth pattern, characteristic of postnatal myocyte growth (also referred to as eutrophy) and of physiological or adaptive hypertrophic growth.
A second type of humoral stimulus involved in cardiomyocyte growth comes from the stimulation of heptahelical G-protein-coupled receptors, particularly the beta 1-adrenergic receptors for epinephrine and norepinephrine, the AT1 receptor for angiotensin II (Ang II) and the ET receptor for endothelin-1, all of which have been related to the development of pathological CH and its eventual progression to heart failure.12,30 In experimental models and clinical syndromes of heart failure, antagonists and synthesis inhibitors of these receptors have modulated the hypertrophic response and improved the prognosis.31 Of these receptors, that most widely studied has been Ang II. In fact, it is now accepted that, together with its regulation of the vasomotor tone and extracellular fluid, the interaction of Ang II with its type 1 receptors (AT1) mediates the response of the myocardial cells to biomechanical stress, involving both those that lead to hypertrophy and those implicated in remodeling.33 It should be pointed out that, while the different heptahelical receptor agonists share a common signaling pattern, there are considerable differences in the cardiac response to these agents, and individual roles for catecholamines versus renin-angiotensin in the cardiac hypertrophic response still remain to be defined.34
Both the cardiomyocytes and the blood vessels possess mineralocorticoid receptors, the stimulation of which can activate important physiological and pathological mechanisms. Peripheral infusion of aldosterone in rats receiving a sodium-rich diet leads to cardiac hypertrophy and fibrosis, an effect that is independent of the increased arterial pressure.35 The mechanism responsible for CH secondary to aldosterone administration is being investigated. Aldosterone has been reported to increase AT1 receptor density, involving both the mRNA and cardiovascular tissue protein,36 and that treatment with losartan partially prevents aldosterone-induced CH and myocardial fibrosis in rats.37 On the other hand, aldosterone also appears to be related to an increase in the activity of the phosphatase, calcineurin, which, as will be dealt with later on, is implicated in the hypertrophic process; the treatment of rats with FK506, a calcineurin inhibitor, partially prevents the prohypertrophic effects of aldosterone.37 Moreover, a number of studies indicate that aldosterone has a direct profibrotic effect, which would be related to the activation of inflammatory mediators and necrotic changes.38-40 These events appear to be responsible, at least in part, for the clinical effect of eplerenone, a selective mineralocorticoid receptor antagonist, in patients with heart failure and left ventricular dysfunction secondary to acute myocardial infarction.41
Inflammatory cytokines have been the object of increasing interest in the field of cardiovascular research. Through their local action, they play a role not only in the pathogenesis of atherosclerosis, but in the cardiac dysfunction that accompanies systemic sepsis, viral myocarditis and other diseases, as well.42,43
One of the cytokines that has received the most attention is interleukin-6 (IL-6) (also known as cardiotropin 1), a 185-amino acid polypeptide that has a number of biological functions, both proinflammatory and antiinflammatory, mediated by its binding to its receptor (IL-6R) and to glycoprotein 130 (gp 130).44 Results relating IL-6 to the development of CH have been published for a number of years. In this regard, it has been reported that neonatal cardiomyocytes cultured in vitro increase in size when exposed to increasing doses of IL-6, and that overactivation of the gp 130 signaling pathway leads to CH in mice.45 These findings have been extended in later works in which an increase in IL-6 expression has been reported in the myocardium of patients with CH who died from myocardial infarction, an increase which would appear to be related to the pathogenesis of the observed hypertrophy.46
It is essential to stress the fact that, here, the different stimuli have been described separately for explanatory reasons, but that this separation does not exist in the real pathophysiological situation, in which there is an intricate network of interconnections among these elements, a circumstance that complicates their study considerably and requires caution when interpreting the reported phenomena. On the other hand, we should not overlook the fact that two thirds of the cells of the normal heart are nonmuscle cells, mostly fibroblasts,47,48 whose role in the pathogenesis of several pathological processes, such as CH and myocardial remodeling, appears to be crucial.49,50 In response to different types of stress, these cells present phenotypic changes, with the expression of markers characteristic of smooth muscle cells. For this reason, they are referred to as myofibroblasts, which produce and release substances such as growth factors, cytokines, extracellular matrix (ECM) proteins and proteases.51,52 Both fibroblasts and myofibroblasts play an important role in regulating the synthesis and degradation of the ECM, the composition and amount of which is the result of a fine balance between collagen synthesis (and, to a lesser extent, the synthesis of other proteins) and the activity of metalloproteases and tissue inhibitors of metalloproteases.53 The disturbance of this delicate balance can lead to fibrotic changes that, together with CH, fibroblast proliferation and cell death, constitute the phenomenon of remodeling.54
The "dialogue" between fibroblasts and cardiomyocytes appears to play a role in both the physiological situation and the pathogenesis of CH and other diseases. For example, the mechanical stretch of cardiomyocytes induces Ang II release which, in turn, provokes the release of a number of growth factors and cytokines on the part of fibroblasts and myofibroblasts, which affect themselves through autocrine mechanisms, and other cell types, including cardiomyocytes, in a paracrine manner. Thus, the available evidence indicates that, just as the paracrine agents released by cardiac fibroblasts are relevant in the induction of the hypertrophic response of the cardiomyocyte, the paracrine activities of the latter are also important for the regulation of fibroblast function.55,56 Moreover, the heart has other cell types, like endothelial cells, pericytes, smooth muscle cells and immune system cells, which also participate in this communications network and probably play a role in the pathogenesis of myocardial remodeling, which, in turn, can affect the course and progression of CH to heart failure.57
Intracellular Signaling in the Hypertrophic Cardiomyocyte
Signal transduction in response to mechanical stress is characterized by the multiplicity of pathways that are activated simultaneously.58 Thus, in cardiomyocytes in culture, mechanical stress produces the activation of phospholipases C, D and A2 (PLC, PLD and PLA2), tyrosine kinases, p21ras, Raf-1, mitogen-activated protein kinases (MAPK) and their activators, JNK kinases, protein kinase C (PKC), and probably other molecules.6,8 This is one of the subjects that has received the most attention in recent years and, with the new advances, the already large number of second messengers that are known to take part in the process can be expected to increase.
Mechanical stress in cardiomyocytes activates PLC, as well as PLD and PLA2, which generates several lipid-derived second messengers, such as inositol 1,4,5-triphosphate (IP3), diacylglycerol (DAG), arachidonic acid, and phosphatidic acid.8 Several possible mechanisms and intermediaries have been proposed for phospholipase activation in response to mechanical stimulation, especially c-src, Ang II,24 and the increase in intracellular calcium through the activation of calcium-permeable and gadolinium-sensitive ion channels.59 While these alternatives are highly appealing, none of them has been shown to really play a role as an intermediary between membrane stretch and phospholipase activation.12 It has also been pointed out that the mechanical stimulus could favor the accessibility of membrane-associated phospholipases to their substrate. In lipid bilayer systems, PLC activity can be increased by the increment in pressure on the monolayer, which expands the space between phospholipids and permits access of phospholipases to the hydrophobic portion of its substrates.58 While this hypothesis is highly interesting, to date, there is no solid evidence to support it.
PLC activation leads to a release of IP3 (which, in turn, produces calcium release from intracellular deposits) and of DAG, which ultimately leads to PKC activation, an event that plays an important role in the expression of immediate early genes, such as c-fos and Egr-1, in response to mechanical stress.6 Of the different isoforms of PKC, those most functionally relevant in the mammalian heart are PKC αand β (belonging to the conventional group and activated in response to an increase in intracellular calcium and DAG) and PKC δ and ε (which are calcium-independent but are activated by DAG).60 The role of these isoforms in the pathogenesis of adaptive and nonadaptive CH in the human heart is not entirely clear, and is a matter of discussion owing to the functional redundancy among the distinct isoforms and to the interspecies variation that has been detected in their expression and activity.34 Despite these limitations, the current consensus is that, of these isoforms, PKC β is probably that which has the most significant role in nonadaptive CH since the remainder would have functions related to the regulation of phenomena such as myocardial contractility, response to ischemia and/or physiological cardiomyocyte growth, participating less in the pathogenesis of pathological hypertrophy.61-64
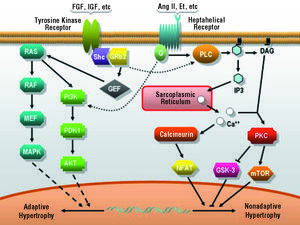
There is evidence that the stimulation of myocyte growth, as well as environmental stresses, including ultraviolet radiation, osmotic stress and DNA damage, activate protein kinase cascades which, in turn, activate MAPK, such as ERK, JNK, and p38.65 These kinases are serine-threonine kinases and phosphorylate a number of important cellular substrates for cell growth and differentiation processes, including CH. In recent years, the contribution of the different protein kinase cascades to the events involved in the development of adaptive or pathological CH has been the subject of fairly detailed study. A growing body of evidence supports the notion that the pathway composed of I αsubgroup of phosphoinositol-3-kinase (PI3K α), phosphoinositol-dependent kinase (PDK1), and the protein kinase Akt (also known as PKB), characteristically activated in response to growth factors such as growth hormone and insulin, participates in the intracellular signal transduction of adaptive or physiological CH66-69 (Figure 2). It is interesting to note that the myocardial hypertrophy observed in mice overexpressing an active form of PI3K α did not progress to nonadaptive hypertrophy whereas, in contrast, the cardiac expression of a dominant negative mutant form of PI3K* limited normal cardiac growth in the animals and prevented the development of physiological hypertrophy in response to exercise.66

Figure 2. Major pathways activated in response to hypertrophic stimuli. Ang II indicates angiotensin II; DAG, diacylglycerol; Et, endothelin; FGF, fibroblast growth factor; G, G protein; IGF, insulin-like growth factor; IP3, inositol 1,4,5-triphosphate; MAPK, mitogen-activated protein kinase; MEK, MAP kinase kinase; NFAT, nuclear factor of activated T cells; PKC, protein kinase C; PLC, phospholipase C; RAF, mitogen-activated protein (MAP) kinase kinase kinase; RAS, monomeric GTPase.
On the other hand, the stimulation of G-protein-coupled heptahelical receptors by the binding of angiotensin, endothelin, catecholamines and other neurohormones leads to PKC activation and to the release of calcium from intracellular deposits, with parallel activation of calcineurin phosphatase and of nuclear factor of activated T cells (NFAT). It should be pointed out that CH associated with the overexpression of the Gq protein in the mouse heart corresponds to eccentric hypertrophy and, despite the fact that the baseline ventricular function was within normal limits, the contractility of individual ventricular cardiomyocytes was reduced; moreover, under certain types of genetic and environmental stress, the animals rapidly developed heart failure. Together, these phenomena indicate that this growth could correspond to nonadaptive CH and, in turn, provides certain clues with respect to the mechanisms causing the transition between hypertrophy and failure.70-72
Despite this suggestive and appealing evidence supporting different signaling pathways for the physiological and pathological hypertrophic processes, we should stress the fact that, in the cellular context, there is extensive interconnection among cascades and second messengers; thus, the phenomena described here should be interpreted carefully and with this circumstance in mind34 (Figure 2).
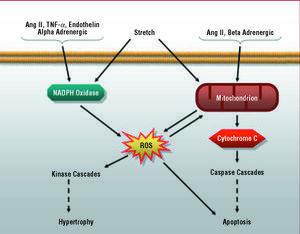
In recent years, increasing numbers of studies have reported an increment in reactive oxygen species (ROS) in the myocardium of patients and animals with heart failure,73-75 a fact that could be due to 2 possible mechanisms: a reduced antioxidant capacity or increased ROS production. Although the first is controversial,76 consensus has been reached regarding the fact that heart failure is accompanied by an increase in free radicals, most of which would originate in cardiomyocyte mitochondria.77 However, there are also cytoplasmic sources of ROS, particularly the cellular oxidative complexes such as NADPH oxidase, xanthine oxidase and nitric oxide synthase.78 A number of groups have reported that Ang II, endothelin-1, TNF- α and alpha-adrenergic agonists produce cardiomyocyte hypertrophy though a ROS-dependent pathway.73 While the mechanism by which free radicals are involved in the action of these agonists is not known in detail, there is a significant body of evidence indicating the participation of the NADPH oxidase complex in the process. Indeed, in contrast to what occurs in immune system cells, in which NADPH oxidases produce large quantities of superoxide anion,79 in the cardiomyocyte, these complexes produce small amounts of these compounds, which act as second messengers in the signaling pathways involved in cell growth, including the hypertrophic response80 (Figure 3). In this respect, the administration of an inhibitor of the NADPH oxidase complex in rat cardiomyocytes has been reported to totally prevent ERK1 and ERK2 activation in response to the administration of endothelin-1 or alpha-adrenergic agonists.81

Figure 3. Role of reactive oxygen species (ROS) in intracellular signaling in the cardiomyocyte.
Another aspect that has received considerable attention in the cardiovascular research setting is the apparent role of monomeric GTPases in different pathological processes.82 Some of this interest is the result of recent evidence concerning the relationship of hypolipidemic agents of the statin class (HMG-CoA reductase inhibitors) to the inhibition of the signaling of this protein family.83-85 Ras, the first member of this family to be related to the pathogenesis of CH, induces a significant increase in myocardial mass when an active mutant form is overexpressed in the transgenic mouse heart.86 In agreement with this finding, the expression of this mutant form in neonatal rat cardiomyocytes results in a hypertrophic gene expression,87 while dominant negative Ras mutants block the phenylephrine-dependent increases in cell size and protein synthesis.88
The Rho subfamily of Ras monomeric GTPases, which includes RhoA, Rac1 and Cdc42, has been an object of interest in recent years because, in addition to its participation in the pathogenesis of hypertension, it appears to play a relevant role in the signaling process that leads to CH.89 RhoA activates several protein kinases, such as ROCK (Rho-associated kinase), and potentiates the activity of the transcription factor, GATA-4, inducing the hypertrophic phenotype in neonatal rat cardiomyocytes.90 The overexpression of RhoA in transgenic mouse myocardium, however, is not enough to induce a hypertrophic response in the ventricle; in contrast, it leads to conduction abnormalities, bradycardia, cavity dilation and heart failure.91 A novel protein referred to as a "striated muscle activator of Rho signaling" (STARS) has recently been described. It binds to the I-band of the sarcomere of cardiac and skeletal myocytes of different species and is capable of RhoA activation actin polymerization which, in turn, leads to changes in transcription factor activity.92 This is interesting since it could lead us to speculate that this pathway could constitute a bridge between the mechanical changes in the sarcomere and gene expression.
Na+/H+ exchanger activity has been reported to be increased in several in vivo and in vitro models of CH.93 This phenomenon leads to a depletion of the transmembrane Na+ gradient, which results in a Na+/H+ exchanger-mediated increase in Ca++ and the activation of several signaling cascades.94 Consistent with this finding, Na+/H+ exchanger inhibition by the specific inhibitor, cariporide, has proved to enhance salvage in different of in vivo models of CH.95,96 Taking into account the fact that the inhibition of this pathway does not appear to have deleterious hemodynamic consequences, some authors have proposed this approach as an eventual target for antihypertrophic therapies.2 Quite a lot of attention has also been focused on changes in the Ca++ cycle in CH. Heart failure is associated with a minor increase in intracellular Ca++ during systole, due, at least in part, to the lower reserves of this ion. This depletion, in turn, is the consequence of adrenergic activation, of the reduced expression of SERCA2a (sarcoplasmic reticulum Ca++ pump) and of increased phospholamban phosphorylation, a circumstance that augments the tonic inhibitory action in SERCA2a. Interesting therapeutic approaches have focused on restoring Ca++ concentrations in the sarcoplasmic reticulum, whether by decreasing the activity of the alpha-adrenergic receptor, by SERCA2a overexpression or by the elimination of phospholamban.98
There is a wide range of agents that also appear to play a role in signal transduction during the hypertrophic process, including the IL-6 family and its receptors,43 calcineurin phosphatase and NFAT,99 growth factors (FGF, TGF-β, IGF-1)12 and the actin cycle.100 There is a great deal of evidence for and against each of them and, at the present time, they are the subject of intense research since several of them, in addition to being implicated in the pathogenesis of CH, seem to have central roles in other pathological processes.
Gene Expression in the Hypertrophic Cardiomyocyte
The intracellular signaling triggered by the different mediators induced by the hypertrophic stimulus are ultimately translated into messages that gain access to the cardiomyocyte nucleus, a process that, in turn, leads to changes in the transcription of numerous genes. In contrast to what occurs with the mechanisms controlling gene expression in skeletal muscle, which have been studied in considerable detail, the processes underlying gene control in smooth muscle and cardiac muscle are still poorly defined.101
It is known that, regardless of the species, the cardiomyocyte responds to the pathological hypertrophic stimulus with a characteristic succession of changes in gene expression. The first group of genes to be activated during mechanical stress are the early response genes, such as c-fos, c-jun, and c-myc, the transcription of which takes place 30 minutes later and peaks within the first hour.102 There is evidence indicating that cardiac expression of the c-fos and c-myc genes is activated in rats with aortic constriction102,103 and that c-fos expression is proportional to the mechanical stretch experienced by the ventricular wall in hearts subjected to extracorporeal perfusion.104 The majority of these genes are transiently overtranscribed, but the transcription normalizes a few hours later, even though the stimulus persists.7 Apparently, this transient response corresponds to a general pattern of induction of growth in cells that have lost their capacity to replicate their genetic material.32
Moreover, CH is associated with over-regulation of the fetal program, with ANF, β MHC and SKA genes, among others.105-107 The expression of these genes is delayed, usually taking place within 6 to 12 hours after the production of the stimulus. They require activation of protein synthesis105 and encode contractile proteins that are normally present in embryonic cardiomyocytes and reexpressed during CH.102,106,107 A number of regulatory elements have been identified that can induce the expression of such fetal genes in response to alpha adrenergic agents or endothelin-1. However, these regulatory elements have not been fully characterized in the case of mechanical stress.103 A large number of experiments with reporter gene constructs under the control of some of these fetal gene promoters have demonstrated little or no response to stretch.108,109 It has been argued that this could be the consequence of methodological errors: in these experiments, short promoters would be been used, which would deprive the artificial construct of the domain capable of responding to mechanical stress.8 However, most researchers agree that these results are valid and appear to indicate that the signaling induced by cell stretch may involve a distinct and particular mechanism.8
The agents causing the changes in gene expression are proteins capable of binding to certain DNA sequences referred to as transcription factors. In the case of cardiovascular tissue, these factors act as regulators of cardiac gene expression that encode structural or functional proteins characteristic of cardiomyocytes. A growing number of these agents have been described in recent years, and the in-depth knowledge of the promoter regions essential for gene transcription, and of the identity of the different transcription factors implicated in the activation of said promoter signals, will be scientific challenges in cardiovascular research for the coming years and a requirement for the design of future conventional and gene therapies. Among these factors, of particular interest are GATA-4, GATA-5, GATA-6,110 Nkx2,5,111, TEF-1,112 MEF-2, myocardin,112,113 SRF,114-116 HOP,113,117 Sp,118,119 hCB1, and hCB2120 and HAND.121 It is interesting that many of these agents present interactions with one another, both synergistic and antagonistic, a circumstance that undoubtedly contributes to making this intricate process even more complex.
During the progression of pathological CH in animal and human models, the heart undergoes a change in metabolism: the major source of ATP generation, which, under normal condtions, is fatty acid oxidation, comes to be glucose utilization.122,123 The so-called PPAR (peroxisome proliferator-activated receptors) correspond to a group of transcription factors that have recently been the object of a great deal of interest, given their clinical importance and the central role they play in the regulation of lipid metabolism in different species, including humans.124,125 The 3 known isoforms (α β, and γ) are expressed in the mammalian heart. They become activated in response to fatty acids, binding like heterodimers to the retinoic acid receptor (RXR) and to consensus sequences in the regulatory regions of a number of genes.126 Recent evidence has related the deactivation of the PPAR α signaling pathway to the reduced gene expression of the enzymes involved in fatty acid oxidation in the hypertrophic heart.127,128 An interesting aspect is the fact that, in rats treated with fenofibrate, a PPAR γagonist, an inhibition of CH secondary to pressure overload was observed,129 a finding that has been expanded on in studies in hypertensive patients with CH.130 The γ isoform of PPAR, on the other hand, not only appears to play a protective role in CH, but also in hypertension-related vascular remodeling which, at least in part, would be mediated by an increased release of vasodilator substances, such as nitric oxide.131-133 The hypertrophic effect of PPAR α and γ is probably related to the regulation of the fatty acids available for uptake by the myocardium and to an acceleration of their oxidation, which results in an increase in ATP synthesis.134,135 This subject has been addressed in detail in recent publications.136,137
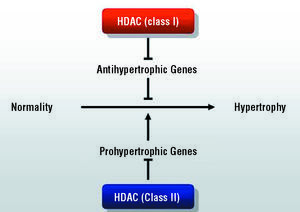
Another particularly important phenomenon involved in gene expression is histone acetylation. This reaction, mediated by histone acetyl transferase (HAT), modifies chromatin structure, which results in its decondensation and transcription activation. The reverse reaction is catalyzed by histone deacetylases (HDAC) which, through deacetylation, induce a compacting of the chromatin and repression of transcription.138-140 Thus, the balance between HAT and HDAC seems to play an especially important role in the control of gene expression, which, according to recent evidence, will also affect the hypertrophic process in cardiomyocytes.141-143 At least 9 mammalian HDAC genes have been described, and are grouped into two major categories: class I and class II. According to available evidence, these enzymes appear to play opposing roles in the hypertrophic process. Thus, whereas the role of HDAC I would be to inhibit antihypertrophic genes and, therefore, would favor myocyte growth, the action of HDAC II would be to "sequester" the transcription factor MEF-2 and, thus, would inhibit the hypertrophic phenomenon144 (Figure 4). Although this line of research is yet to be explored in detail, it has aroused a great deal of interest because of its potential clinical applications, which are not limited to the area of cardiovascular diseases. Indeed, chemical inhibitors of HDAC have been identified and synthesized, and recent studies indicate that these compounds, which have demonstrated their potential use as antineoplastic drugs,145 would have a role in promoting differentiation in myeloid tumors. These drugs are surprisingly well tolerated and, at the present time, they are being assessed in several ongoing phase I and phase II trials.146

Figure 4. Model of the role of the 2 classes of histone deacetylases (HDAC) in the pathogenesis of cardiomyocyte hypertrophy.
Microarray technology has recently been applied to the identification of genes that are altered in CH. For this purpose, a mouse model of drug-induced hypertrophy (isoproterenol or Ang II) was utilized, and the expression profile of more than 4000 genes was analyzed during the induction and regression of the hypertrophic response. The participation of 25 genes or pathways previously reported to be relevant in the hypertrophic process was confirmed, but, in addition, 30 genes that had not previously been related to cardiomyocyte hypertrophy were identified. Of the 55 genes that undergo an overall change of expression, 32 were altered only during induction and eight during regression, whereas the remaining 15 exhibited changes of expression during both phases.147 If we take into account the fact that this study focused on only 3% of the mouse genome, it can be expected that many other genes that are relevant in the hypertrophic process have yet to be identified, a circumstance that indicates the complexity of the events taking place here, and the limited extent of our knowledge in this respect.
The regulation of protein synthesis represents a crucial point in all forms of CH. Two components can be distinguished in this process: the overall control of protein synthesis and the regulation of specific mRNA. In turn, the overall control of protein synthesis involves ribosome biogenesis and activation of the translation machinery.34 Both ribosome biogenesis and activation of the translation machinery depend, at least in part, on the protein kinase, mTOR. Both growth factors and neurohormonal factors can lead to mTOR activation in mammalian cells but, while the former do it through PI3K*, substances like Ang II, endothelin or norepinephrine lead to the activation of this protein through PI3K* and the MAPK pathway. According to this model, mTOR could represent a final common pathway for the different signals leading to cardiomyocyte hypertrophic growth. In support of this hypothesis, it has been shown that rapamycin, an inhibitor of mTOR, blocks and can even reverse CH secondary to pressure overload.66 Among the effectors of the hypertrophic response triggered by mTOR, of particular interest are the protein kinases S61 and S62, central regulators of ribosome biogenesis, translation, cell cycle progression and hypertrophy,148,149 and regulatory factors of the translation machinery, such as the translation initiation factor, eIF4E, and the translation elongation factor, eEF2, both of which are disinhibited in response to the action of mTOR and permit translation35 (Figure 2).
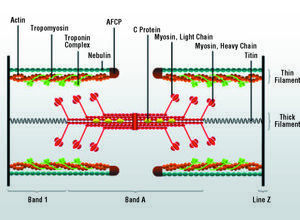
The disposition of the synthesized proteins plays an important role in myocyte growth, especially in the so-called process of sarcomerogenesis, which leads to the formation of the complex protein structure corresponding to the sarcomere (Figure 5). While the linear addition of sarcomeres leads to longitudinal cardiomyocyte growth, the assembly in parallel increases its transverse diameter, a circumstance that may have important consequences for ventricular architecture and function. The direction of growth is controlled by posttranscriptional mechanisms that are yet to be fully understood.150 In this respect, one of the proposed hypotheses maintains that the control of the addition of new sarcomeres is based on the capacity of the cell to deliver mRNA to specific sites in the contractile apparatus, where they will be translated and assembled in situ. This hypothesis is supported by studies in skeletal and cardiac myocytes, in which a differential localization of cytoskeletal and sarcomeric mRNA has been observed during periods of rapid cell growth and/or intense muscle activity.151,152 It appears, however, that the disposition and assembly of new sarcomeres does not depend exclusively on this local translation mechanism, but responds to a coordinated and regulated series of processes that involves the interaction of multiple proteins and intracellular signals,153-157 many of which are directly or indirectly linked to cardiomyocyte hypertrophic growth.

Figure 5. Structure of the sarcomere with its major proteins. AFCP indicates actin filament capping protein.
CONCLUSION
The reader of these pages will clearly see that the process of pathological myocardial hypertrophy, like most biological phenomena, is a highly complex event involving different cell types, in which an entire range of stimuli, membrane receptors, intracellular signaling cascades, transcription factors, genes and effectors bring about, jointly, changes in the architecture and function of the organ. A scientific approach to this problem not only is possible, but necessary, and constitutes a task of the utmost interest, owing both to its peculiar biological features and to its importance in terms of public health in many countries.
The practical implications of the study of the molecular mechanisms that ultimately cause the hypertrophic program to proceed undoubtedly appear to be far off at the present time. However, we consider that systematic and cooperative work and research in this field can result in yet unsuspected benefits. For many, the promise of a future gene therapy that will make it possible to limit the development of CH and its progression to heart failure continues to be an attainable goal. But the battlefronts are not limited to this possibility alone. Interesting prospects are also emerging in other fields, such as the study of polymorphisms and mutations in transcription factors that account for a greater susceptibility to cardiac hypertrophic stimuli, which will enable us to identify molecular risk factors associated with this disease, and to characterize different segments of the population according to their genetic imprint and lifestyle. This, in turn, would allow us to evaluate our resources better and channel them more efficiently.
Funded in part by grant no. 1030181 from the Spanish Fondo Nacional de Desarrollo Científico y Tecnológico (Fondecyt).
Correspondence:
Dr. J.E. Jalil.
Departamento de Enfermedades Cardiovasculares.
Pontificia Universidad Católica de Chile.
Lira, 85, piso 2.
Santiago, Chile
E-mail: jjalil@med.puc.cl