Myotonic dystrophy type 1 is characterized by muscle damage and systemic manifestations, including cardiac involvement. Our aim was to document the frequency and severity of cardiac involvement (left ventricular dysfunction and arrhythmia or conduction disorders), the need for a pacemaker, implantable cardioverter-defibrillator, or electrophysiological study, and the development of sudden death during follow-up.
MethodsRetrospective observational study of myotonic dystrophy type 1 patients referred to a specialized cardiac unit. Patients received clinical, electrocardiographic (Holter monitoring), and echocardiographic follow-up.
ResultsWe included 81 patients (51.9% men; mean age, 29.9 [14.8] years). The mean follow-up was 5.7 (3.9) years (range: 1-20 years). During this period sinus bradycardia was documented in 48.8%, PR interval≥220 ms in 31.3%, long corrected QT interval in 5%, and QRS interval≥120 ms in 7.5%. A total of 13.8% of patients developed sinus node dysfunction, 10% of patients had supraventricular arrhythmias, 5% had ventricular tachycardia, and 8.8% developed second- or third- degree atrioventricular block. Only 1 patient had severe ventricular dysfunction. During the follow-up, 15 pacemakers and 2 implantable cardioverter-defibrillators were implanted and 5 electrophysiological studies were performed (mainly due to ventricular tachycardia). There was only 1 sudden death.
ConclusionsArrhythmia or conduction disorders are frequent during the course of myotonic dystrophy type 1 patients. A significant percentage of patients require electrophysiological study and the use of a device (pacemaker or implantable cardioverter-defibrillator). In our experience, systolic dysfunction and sudden death are rare.
Keywords
Myotonic dystrophy type 1 or Steinert's disease is currently the most common form of muscular dystrophy in adults. Inheritance of this multisystem disease is autosomal dominant, and phenotypic expression is highly variable due to an unstable CTG trinucleotide repeat in the long arm of chromosome 19 (19q.21.3), which encodes a protein kinase. This disease is associated with myotonia, progressive muscle weakness with atrophy of skeletal muscles, and numerous systemic manifestations, of which the most common are cataracts, diabetes mellitus, hypogammaglobulinemia, mental retardation, and heart disease.1 Respiratory failure (up to 43%) is the most common cause of death, followed by cardiac events (up to 20%).2
Cardiac involvement is due to myocardial fibrosis and degeneration of the conduction system, which also creates a substrate for reentrant arrhythmias and contributes to the ventricular dysfunction that can occur in these patients. Cardiovascular manifestations are therefore mainly characterized by impaired left ventricular function (both systolic and diastolic, although many of these patients do not report clinical heart failure)3 and, more frequently, by electrocardiographic changes (in up to 30.2% of patients).4,5 Other manifestations include prolonged PR and QT intervals, QRS widening, the development of sinus node dysfunction, second- and third-grade atrioventricular block (AVB), right branch bundle block, and left atrial or ventricular tachyarrhythmias.6
Several studies have associated electrocardiographic baseline abnormalities with a risk of sudden death,6 leading to pacemaker (PMK) implantation in 4.1% to 11% of patients and the use of an implantable cardioverter defibrillator (ICD) in 1.1% to 5.3%,7 especially when a Holter study is used.8,9 Risk of sudden death per year of follow-up, or of implantation of a PMK or ICD is currently estimated at 0.56%, 1%, and 0.2%, respectively.8
Given the frequency of rhythm or conduction disorders and the risk of sudden death in these patients, and as there is still no consensus regarding their monitoring,8,9 some authors recommend using invasive diagnostic techniques when abnormalities are found on the baseline electrocardiogram. These diagnostic techniques are recommended even when patients are asymptomatic, the aim being to increase survival.4,10,11 The objective of this study was to document the prevalence and severity of cardiovascular disease in these patients and to analyze the need for PMK, ICD, or electrophysiological study (EPS), and to investigate the occurrence of sudden death during follow-up.
METHODSWe conducted a retrospective observational study of patients with myotonic dystrophy type 1 referred from the neurology department to a specialized cardiology unit at Nuestra Señora de Candelaria University Hospital on the island of Tenerife, between 1983 and 2010. This tertiary center serves a population of 424 923 inhabitants (20 municipalities in the south of Tenerife and the population of the islands of La Gomera and El Hierro). The definitive diagnosis of Steinert's disease was made using a genetic study (polymerase chain reaction, or Southern blot when the phenotype was obvious) performed when there were signs of a neurological condition.
In the first visit to the cardiology unit, a cardiac history was taken and a resting electrocardiogram, Holter monitoring, and echocardiogram were performed. Follow-up consisted of an annual clinical review and electrocardiogram, and further Holter monitoring and echocardiography every 2 years, unless clinical, electrocardiographic abnormalities, or echocardiographic findings indicated that earlier follow-up was required.
We analyzed the systemic manifestations of the disease, family history of sudden death, and the degree of muscle involvement. The latter was assessed using Mathieu et al.’s Muscular Impairment Rating Scale,12 which classifies muscular involvement in 5 categories: 1, no muscle weakness; 2, minimal signs without distal weakness, except in distal flexor muscles of the lower extremities; 3, unaffected proximal distal weakness, except for elbow extensors; 4, moderate proximal weakness, and 5, significant proximal weakness.
Left ventricular ejection fraction was assessed by echocardiography using the Teichholz method. Patients were considered to have ventricular dysfunction if left ventricular ejection fraction was<50%.
Electrocardiographic parameters assessed at baseline were heart rate, PR interval, QRS width, and corrected QT interval (QTc). Sinus bradycardia was defined as sinus rhythm<60 bpm or daytime sinus pauses>3s, and long QTc as>450ms in men and>470ms in women. All Holter studies were reviewed for supraventricular tachyarrhythmias (especially atrial fibrillation or flutter), AVB II or III, sinus node dysfunction (presence of sinus bradycardia and daytime pauses >3s sometimes associated with supraventricular tachycardia) and sustained or nonsustained ventricular tachycardias (VT). We also documented the need for ICD or PMK implantation and EPS, and analyzed patient mortality during follow-up.
Statistical AnalysisThe Kolmogorov-Smirnov test was used to determine the normality of distribution in continuous variables. Categorical variables are presented as n (%) and quantitative variables as mean (standard deviation) if normally distributed, or as median (range) otherwise. The Kaplan-Meier method was used to calculate the probability of PMK-free survival. Analyses were performed using version 15.0 of SPSS.
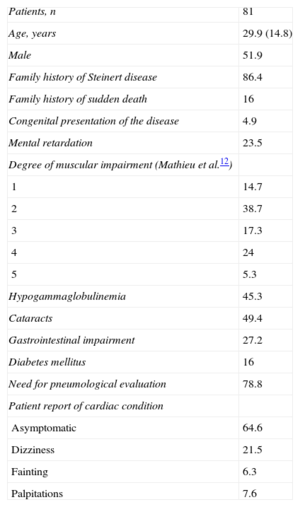
RESULTSDuring the study period, 81 patients with a genetic diagnosis of myotonic dystrophy type 1 were referred to our unit. A total of 51.9% were male and the mean age at diagnosis was 29.9 (14.8) years. Most (86.4%) of the patients had a family history of the disease and 16% had a family history of sudden death. The most frequent neurological condition was grade 2 impairment on the Muscular Impairment Rating Scale (38.7%); cataracts (49.4%) and hypogammaglobulinemia (45.3%) were the most frequent extracardiac conditions. Evaluation by the respiratory unit was required in 78.8% of the patients to determine whether noninvasive ventilation was needed. Table 1 shows the epidemiological and clinical characteristics of the series. During a mean follow-up of 5.7 (3.9) years (range: 1-20 years), 64.6% of patients remained free of cardiovascular symptoms, 21.5% reported dizziness, 6.3% reported syncope, and 7.6% reported palpitations. No patient had symptoms compatible with heart failure and only 1 patient (1.3%) showed severe ventricular dysfunction. There was no evidence of impaired left ventricular ejection fraction in the remaining patients.
Demographic Characteristics and Extracardiac and Clinical Manifestations of Patients Referred With a Genetic Diagnosis of Myotonic Dystrophy Type 1
| Patients, n | 81 |
| Age, years | 29.9 (14.8) |
| Male | 51.9 |
| Family history of Steinert disease | 86.4 |
| Family history of sudden death | 16 |
| Congenital presentation of the disease | 4.9 |
| Mental retardation | 23.5 |
| Degree of muscular impairment (Mathieu et al.12) | |
| 1 | 14.7 |
| 2 | 38.7 |
| 3 | 17.3 |
| 4 | 24 |
| 5 | 5.3 |
| Hypogammaglobulinemia | 45.3 |
| Cataracts | 49.4 |
| Gastrointestinal impairment | 27.2 |
| Diabetes mellitus | 16 |
| Need for pneumological evaluation | 78.8 |
| Patient report of cardiac condition | |
| Asymptomatic | 64.6 |
| Dizziness | 21.5 |
| Fainting | 6.3 |
| Palpitations | 7.6 |
Data are expressed as n (%) or mean±stantard daviation.
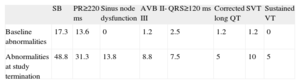
In the first visit, 71.6% of patients had a normal baseline electrocardiogram and sinus bradycardia was documented in 17.3%. The PR interval was≥220ms in 13.6%, second degree Mobitz I AV block was recorded in 1.2%, QRS≥120ms in 2.5%, and long QTc in 1.2%. Supraventricular tachycardia (typical atrial flutter at a normal ventricular rate) was observed in 1 patient (1.2%). At follow-up, only 29 patients (36.7%) had no rhythm or conduction disturbances, while sinus bradycardia was present in 48.8%, PR interval≥220ms in 31.3%, QRS interval≥120ms in 7.5%, and long QTc in 5%. During follow-up, 11 patients also showed sinus dysfunction, 8 had supraventricular tachycardia, 4 had sustained VT, and 7 had grade II-III AV block. Only 7 patients (8.9%) had a baseline abnormality which remained unchanged over the study period and an electrocardiogram with the same characteristics at follow-up as at the first visit. Table 2 shows the electrocardiographic changes in the series over time.
Electrocardiographic Abnormalities Observed During Follow-up in the Study Group (%)
| SB | PR≥220 ms | Sinus node dysfunction | AVB II-III | QRS≥120 ms | Corrected long QT | SVT | Sustained VT | |
| Baseline abnormalities | 17.3 | 13.6 | 0 | 1.2 | 2.5 | 1.2 | 1.2 | 0 |
| Abnormalities at study termination | 48.8 | 31.3 | 13.8 | 8.8 | 7.5 | 5 | 10 | 5 |
AVB, atrioventricular block; SB, sinus bradycardia; SVT, supraventricular tachyarrhythmias; VT, ventricular tachycardia.
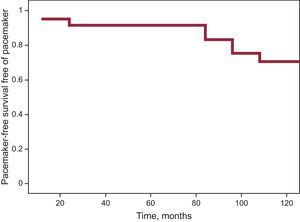
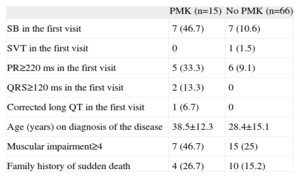
PMK were implanted in 15 patients (20%) during the study period; 93.3% of PMKs implanted were dual chamber. A family history of sudden death, the presence of an advanced muscular condition, and baseline electrocardiographic changes after the first visit were observed more frequently in the group implanted with a PMK. Table 3 shows the characteristics of the group receiving PMKs. Only 1 patient was clinically asymptomatic; 73.3% reported dizziness, 2 (13.3%) had syncope, and 1 had palpitations. The indication for implantation occurred before the electrocardiographic findings were available (sinus node dysfunction in 11 patients, AVB II-III in 3 patients, and alternating bundle branch block observed after EPS, which showed an abnormal HV interval, in 1 patient). Holter recording was a crucial diagnostic tool in 9 patients (documented AVB II-III in 3 patients, alternating and left right bundle branch block in 1 patient, and sinus node dysfunction in the remainder). The estimated probability of PMK-free survival was 83.4%±5.5% at 90 months (Figure).
Electrocardiographic and Clinical Characteristics of Patients Requiring Pacemaker Implantation During their Clinical Course
| PMK (n=15) | No PMK (n=66) | |
| SB in the first visit | 7 (46.7) | 7 (10.6) |
| SVT in the first visit | 0 | 1 (1.5) |
| PR≥220 ms in the first visit | 5 (33.3) | 6 (9.1) |
| QRS≥120 ms in the first visit | 2 (13.3) | 0 |
| Corrected long QT in the first visit | 1 (6.7) | 0 |
| Age (years) on diagnosis of the disease | 38.5±12.3 | 28.4±15.1 |
| Muscular impairment≥4 | 7 (46.7) | 15 (25) |
| Family history of sudden death | 4 (26.7) | 10 (15.2) |
PMK, pacemaker; SB, sinus bradycardia; SVT, supraventricular tachyarrhythmias.
Data are expressed as n (%) or mean±standard deviation.

During follow-up, 2 patients (2.7%) required an ICD: 1 had syncope and severe ventricular dysfunction while the other showed multiple salvos of nonsustained monomorphic VT on Holter recording, despite being asymptomatic. An EPS was performed and induced multiple VT, one of which was ablated. This led to the decision to implant the device.
An EPS was carried out in 5 patients (6.8%): right bundle branch ablation was used to treat bundle branch VT in 3 patients, VT ablation was used in 1 patient, and 1 patient received ablation of the cavotricuspid isthmus to treat typical atrial flutter. The indication for EPS was based primarily on the clinical presentation (2 patients had syncope and 1 had dizziness, with Holter results showing alternating bundle branch block; VT was induced in EPS in all 3 patients). In 2 asymptomatic patients, EPS was indicated by electrocardiographic findings (nonsustained monomorphic VT on Holter monitoring with subsequent induction of VT in 1 and typical atrial flutter in the other).
Eight patients (11%) died during follow-up: 4 from respiratory failure, 1 from colorectal carcinoma which was not susceptible to surgical treatment due to serious muscle weakness, 1 from a head injury sustained in a fall, and another due to lower extremity skin sepsis. Only 1 patient suffered sudden death.
DISCUSSIONThis article describes the clinical course of a group of cardiac patients with Steinert's disease followed in the specialized cardiology unit of a tertiary hospital. Rhythm and conduction disorders were the primary cardiovascular manifestations in these patients, as opposed to the potential development of cardiomyopathy. During a mean follow-up of 5.7 years, most patients showed electrocardiographic abnormalities. While many of these stemmed from the deterioration in the conduction system inherent to the disease, in 20% of patients a PMK implant was deemed necessary. This percentage is higher than the implant rates reported in other series: Breton et al.13 described a PMK implant rate of 3.3% in a cohort of 428 patients with a median age of 33 (2-81) years followed for a mean of 11.7 years, while Lindqvist et al.14 reported an implant rate of 2.7% in 36 patients with a mean age of 45 (10) years followed for a mean of 3 years. The high proportion of patients requiring a PMK in our series was probably due to regular monitoring with a Holter device, which proved to be decisive when deciding on the therapeutic approach in 60% of patients. In a study by Cudia et al.,9 all patients (n=245; mean age, 45 years) were followed using Holter monitoring and a PMK implant rate of 11% was reported.
Given the risk of sudden death in these patients,6 some authors have proposed the use of invasive studies to determine whether changes are required in treatment which may help avoid sudden death.4,10,11,15 In a review by McNally et al.,4 EPS was recommended when baseline electrocardiographic abnormalities are present (PR interval>240ms and QRS interval>120ms); Holter recording was recommended only in asymptomatic patients with a normal baseline electrocardiogram. In our group, patients requiring PMK were diagnosed at an older age than those included in the other studies mentioned9,14 and more often showed electrocardiographic changes after the first visit. These findings probably indicate damage intrinsic to the conduction system preceding sinus node dysfunction (impaired conduction leading to a higher rate of PMK implantation) or advanced AVB. We performed EPS in 5 patients, primarily due to VT, and the main causes of death were related to muscle deterioration. There was only 1 sudden death. The low rate of sudden death during follow-up contrasts with that described by Groh et al.,6 who reported a rate of 33.3% in a group of 406 adults followed for 5.5 years. In that study, an annual electrocardiogram was not a requisite for follow-up, and the need for cardiac evaluation was at the discretion of the general practitioner. In our study, the high rate of PMK implantation was likely instrumental in preventing the occurrence of extreme bradycardia or asystole, which in turn would lead to a lower rate of sudden death. The high rate of device use was influenced by the results of noninvasive monitoring in many patients, with a standard monitoring protocol being applied from the start of follow-up.
Our study also differs from others in the low incidence of ventricular systolic dysfunction. Petri et al.8 analyzed cardiovascular manifestations (cardiomyopathy and rhythm or conduction disturbances) in 18 studies published between 1980 and 2010 and found a prevalence of 7.2% for primary cardiomyopathy. Simply determining the left ventricular ejection fraction by echocardiography is likely to underestimate the true incidence of ventricular dysfunction. For example, Ozyigit et al.3 analyzed transmitral and transtricuspid Doppler flow (standard and tissue Doppler) in 21 patients with myotonic dystrophy type 1 and compared the results with 21 controls. These authors found that an increased Tei index (a myocardial performance tool which provides a noninvasive means to assess alterations in systolic and diastolic function) provided a more sensitive estimate of ventricular dysfunction than left ventricular ejection fraction in these patients. Likewise, Lindqvist et al.14 analyzed 36 patients vs 16 controls using Doppler echocardiography and electrocardiography and concluded that, in the affected group, conventional measures for calculating left ventricular ejection fraction were of little value compared to left ventricle isovolumetric contraction and relaxation times, which they found to be elongated. While these values can be explained by the presence of electrocardiographic abnormalities (lengthened PR interval and QRS interval), in a percentage of patients it is a cause of myocardial rather than electrical asynchrony.
CONCLUSIONSIn our series, most patients with Steinert disease had rhythm and conduction disturbances during follow-up; 20% required a PMK and 2.7%, an ICD. Close electrocardiographic monitoring using a standard protocol is essential and can have a decisive impact on the therapeutic approach in a considerable number of patients. In contrast to rhythm and conduction disturbances, echocardiographic ventricular systolic dysfunction is very uncommon.
CONFLICTS OF INTERESTNone declared.