
“The whole is more than the sum of its parts”
Aristotle“The glory of medicine is that it is constantly moving forward, that there is always more to learn”
Hypertrophic cardiomyopathy (HCM) is an important cause of disability and death in patients of all ages, but sudden, unexpected death in young adults is the most frightful component of its natural history. It is mainly caused by mutations in sarcomeric genes but other factors can modulate the phenotype; in fact, many pathogenic/causative mutation-carriers remain disease-free.
The aim of the elegant study by Pérez-Sánchez et al.1 recently published in Revista Española de Cardiología was to determine whether factors such as sex, systemic hypertension, and physical activity are modifiers of the severity of the disease, and to establish their role in the age-related penetrance of HCM, or prevalence of disease-cases among causative mutation-carriers, in a large cohort of genotyped HCM families with mutations in the sarcomere genes MYBPC3 and MHY7, the most common causative mutations. This cohort enabled detection of differences in phenotype but avoided including patients with multiple mutations causing more unfavorable phenotypes. Reduced penetrance is likely to accrue from combinations of a variety of interacting genetic and external and internal (Bernardian) environmental factors.2
HCM is the most widespread professedly monogenic genetic cardiac disease with an overall prevalence of 0.2% (≈ 700000 cases in the United States), but the prevalence of specific pathogenic mutations is unknown. The discovery of sarcomere mutations as the genetic basis of HCM has provided major gateways into its pathogenesis at the molecular level, including contributions of mitochondrial dysfunction with accompanying energetic abnormalities.
Histopathologically, HCM is a myocardial disease presenting expanded interstitial matrix, enlarged myocyte nuclei, myofiber thickening and misalignment/disarray, and a primary, usually asymmetric, left ventricular wall-hypertrophy (LVH) including papillary muscles and trabeculae. The LVH is a direct sequel of HCM mutations and not compensatory/secondary to a pressure overload.3 It is associated with confined left ventricular (LV) cavity size, but end-stage disease exhibits heart failure and ventriculoatrial dilatation, with wall-thinning and scarring.
MULTISENSOR CATHETERIZATION-PHENOTYPE OF HCMWith multisensor micromanometric/velocimetric catheterization alongside digital cardiac imaging modalities, novel extensive quantitative genotype-phenotype hemodynamic correlations (genome-wide association studies [GWAS]) of HCM are feasible. A predictable relation between genotype and HCM disease-expression is needed to apply genetic testing to clinical decision-making.4 Multimodal studies currently offer the prospect of identifying multifaceted phenotypic HCM changes in systolic and diastolic dynamics,3 when unceasing knowledge advancement is more imperative than in Mayo's time (Epigram).
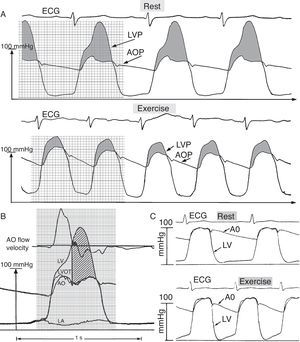
Hyperdynamic systolic and impaired diastolic dynamics are spectacular catheterization HCM hallmarks, especially during exercise, as summarized in Figure 1. HCM exhibits what I have previously called “polymorphic pressure gradients.”3,5,6 Notwithstanding clinical guidelines not recommending intense/competitive exercise, the association between physical activity and outcomes in HCM appears weak.1
 and aortic root (AOP) pressures in hypertrophic cardiomyopathy at rest and during supine bicycle exercise, which elicits an abnormal LVP diastolic decay, suggesting impaired ventricular relaxation; LVP decays throughout diastole, in sharp contrast to the normal pattern shown in panel C. B: Pressure-flow relation with large early and enormous mid- and late-systolic dynamic gradients in hypertrophic cardiomyopathy. From top downward: aortic velocity signal, and deep left ventricular (LV), left ventricular outflow tract (LVOT), and aortic root (AO) micromanometric signals, measured by retrograde triple-tip pressure plus velocity multisensor left-heart catheter. Left atrial (LA) micromanometric signal was measured simultaneously by trans-septal catheter. The vertical straight line identifies the onset of SAM-septal contact, determined from a simultaneous M-mode mitral valve echocardiogram (not shown); the majority of aortic ejection flow is already completed by this time. The huge mid- and late- systolic gradient (hatched area) is maintained in the face of minuscule remaining forward or even negative aortic velocities. SAM, systolic anterior motion of the mitral valve. Adapted from Pasipoularides,3 with permission of PMPH-USA.")
A: Deep left ventricular (LVP) and aortic root (AOP) pressures in hypertrophic cardiomyopathy at rest and during supine bicycle exercise, which elicits an abnormal LVP diastolic decay, suggesting impaired ventricular relaxation; LVP decays throughout diastole, in sharp contrast to the normal pattern shown in panel C. B: Pressure-flow relation with large early and enormous mid- and late-systolic dynamic gradients in hypertrophic cardiomyopathy. From top downward: aortic velocity signal, and deep left ventricular (LV), left ventricular outflow tract (LVOT), and aortic root (AO) micromanometric signals, measured by retrograde triple-tip pressure plus velocity multisensor left-heart catheter. Left atrial (LA) micromanometric signal was measured simultaneously by trans-septal catheter. The vertical straight line identifies the onset of SAM-septal contact, determined from a simultaneous M-mode mitral valve echocardiogram (not shown); the majority of aortic ejection flow is already completed by this time. The huge mid- and late- systolic gradient (hatched area) is maintained in the face of minuscule remaining forward or even negative aortic velocities. SAM, systolic anterior motion of the mitral valve. Adapted from Pasipoularides,3 with permission of PMPH-USA.
I have developed a flow-model that elucidates late-systolic HCM hemodynamics with cavity elimination.3,5,6 Whether mitral leaflet-septal contact causes outflow obstruction and the enormous mid- and late-systolic intraventricular gradient remains controversial (Figure 1B). This is recounted by Braunwald,7 the main HCM pioneer under whose inspired leadership I had the privilege, early in my career, to work in the Department of Medicine at the Peter Bent Brigham Hospital, at Harvard.
Coexisting prominent biventricular diastolic function abnormalities are also demonstrated in Figure 1, especially during acute physical exercise.3,8 The interacting factors3 that underlie LV inflow dynamics9 and the pronounced heterogeneity in the relative contributions of relaxation defects,10 asynchrony,11 altered passive diastolic properties and geometry12,13 to the overall diastolic dysfunction in HCM, require further investigation.
GENETIC/GENOMIC CONTEXT OF HCMHCM involves disease-causing genetic variants and exhibits morphological, functional, clinical and prognostic inter- and intrafamilial phenotypic heterogeneity. However, intrafamilial inconsistencies are not explained by mutational heterogeneity; accordingly, environmental factors must be implicated too. Reduced/incomplete penetrance and variable expressivity (phenotypic plasticity) of pathogenic/causative mutation(s) have impeded an across-the-board understanding of its clinical spectrum. Phenotypic diversity among individuals ensues from variations in DNA sequence and from internal/external environmental influences on genes with variable expressivity/incomplete penetrance.3 A combination of genetic, environmental, random chance, and lifestyle factors render the probability that HCM-gene carriers will manifest the disease phenotype < 1.
The sequencing of the human genome has allowed definitive linkage of a growing number of diseases including HCM to numerous genetic/genomic polymorphic variants, as summarized in the iconic United States National Human Genome Research Institute-European Bioinformatics Institute (NHGRI-EBI) GWAS Catalog in Figure 2. The diagram is released nightly and the latest version is made available on the NHGRI-EBI Catalog website.14 The majority of HCM mutations are “private,” ie, family-specific. HCM is caused by mutations in genes encoding mainly sarcomeric contractile apparatus proteins15; most are missense mutations consisting of a replacement of an amino acid by another. These modify physical/functional properties of proteins incorporated in sarcomeres and may induce hypertrophic signals. Less commonly, insertions or deletions of nucleotides arise in frameshift mutations, which profoundly alter messenger RNA translation and resultant protein properties.
 Catalog is supplied jointly by the National Human Genome Research Institute (NHGRI) and the European Bioinformatics Institute (EMBL-EBI). The diagram is released nightly and the latest version is made available on the NHGRI-EBI Catalog website.14")
Diagram showing all single nucleotide polymorphism-trait associations with P-value ≤ 5.0 × 10-8, mapped onto the human genome by chromosomal locations and displayed on the human karyotype. The Genome-wide Association Studies (GWAS) Catalog is supplied jointly by the National Human Genome Research Institute (NHGRI) and the European Bioinformatics Institute (EMBL-EBI). The diagram is released nightly and the latest version is made available on the NHGRI-EBI Catalog website.14
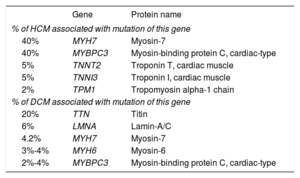
By inference from currently commercially available HCM multigene panel testing lists,16 up to now, pathogenic/causative mutations have been identified in ≈25–30 genes, including MYBPC3, MYH7, TNNI3, TNNT2, TPM, MYL2, MYL3, ACTC, ACTN2, and TCAP. Notably, MYH7 and MYBPC3 gene mutations account for ≈ 80% of genotyped cases, as shown in the Table.15,17
Genes Commonly Implicated in Assorted Cardiomyopathies in Descending Order of Frequency
| Gene | Protein name | |
|---|---|---|
| % of HCM associated with mutation of this gene | ||
| 40% | MYH7 | Myosin-7 |
| 40% | MYBPC3 | Myosin-binding protein C, cardiac-type |
| 5% | TNNT2 | Troponin T, cardiac muscle |
| 5% | TNNI3 | Troponin I, cardiac muscle |
| 2% | TPM1 | Tropomyosin alpha-1 chain |
| % of DCM associated with mutation of this gene | ||
| 20% | TTN | Titin |
| 6% | LMNA | Lamin-A/C |
| 4.2% | MYH7 | Myosin-7 |
| 3%-4% | MYH6 | Myosin-6 |
| 2%-4% | MYBPC3 | Myosin-binding protein C, cardiac-type |
These genes encode proteins of the sarcomere, Z-disc and calcium regulation (Ca2+ signaling is an essential trigger of hypertrophy) with varying pathogenicity. They all intensify Ca2+ sensitivity/signaling and myofilament function bringing about myocyte hypercontractility, which increases sarcomere force generation and actin-myosin sliding velocities. It begets enhanced adenosine triphosphate (ATP) hydrolysis and a negative energy balance; given time, diastolic dysfunction will ensue.3
That an energy deficit is essential in causing the HCM phenotype is suggested by several inherited syndromes with impaired mitochondrial energy production and asymmetric cardiac hypertrophy clinically indistinguishable from HCM. Gene variants/alleles yielding such HCM phenocopies (ie, traits typifying another genotype) include those of Fabry disease (GLA), Danon disease (LAMP2), Pompe disease (GAA), glycogen storage disease (PRKAG2), and others linked to mitochondrial respiratory chain defects (referred to above). Genetic testing distinguishes these from HCM.
Abnormal Ca2+–ATP Relationships Underlie HCM Pathophysiology/PathologyAs discussed above, the Ca2+ sensitization of HCM-linked mutations raises systolic contractility, but with an ensuing Ca2+ dysregulation that causes impaired cardiomyocyte relaxation, accruing from slowed cross-bridge cycling rates and therefore directly from HCM mutations. Relaxation/diastolic dysfunction should contribute to the incidence of sudden death in HCM, due either to lethal ventricular arrhythmias or to abnormally high cytosolic free Ca2+. Such Ca2+ elevations are deleterious, partly because overloading mitochondria with Ca2+ causes a decrease in ATP formation due to depolarization of the inner mitochondrial membrane.18 Markedly reduced mitochondrial ATP generation prevents Ca2+ pumps, in the sarcolemma and the sarcoplasmic reticulum network around each myofibril, from clearing the cytosol of the excessive Ca2+, eliciting a vicious circle. This should likely be exacerbated by any exercise-/stress-induced augmentation of cardiac sympathetic drive.
Additionally, elevated cytosolic free Ca2+, particularly during diastole, would also heighten cardiomyocyte hypertrophy through activation of various Ca2+-dependent gene expression-changing signaling pathways, including calcineurin/NFAT/GATA4 and/or calmodulin-dependent kinase II.,19–21 The ensuing relative/absolute ATP depletion worsens the impaired sarcoplasmic reticulum Ca2+ reuptake and diastolic dysfunction.3,12
HCM IN THE GENOMICS ERA: GENOTYPIC AND PHENOTYPIC VARIABILITYThe genetics and biology underlying HCM is presently at an exciting inflection point. The rapid innovations in genotyping technologies that have occurred over the past several years have enabled GWAS for finding additional HCM candidate genes to correlate with disease susceptibility.15
Deciphering the functional/morphomechanical impacts of polymorphic variants in the human genome entails ascertaining their effects on gene expression. However, active mutation expression likely fluctuates over time. Genes can be regulated, ie, turned on-and-off at apposite times and in appropriate contexts by particular cues/signals coming from outside the genome and interacting with controlling promoter, operator, and silencer DNA “switches” located alongside the genes. This regulation employs transcription factors, proteins that bind to DNA at certain target sequences, rendering it harder or easier for RNA polymerase to bind to the promoter of the gene and begin transcription. They act like rheostats—if one slides the controller up-or-down, one gets more or less of a product/effect. This process generally allows cells to perform logic operations and combine different sources of ever-changing information to “decide” whether to express a gene; it ordinarily allows genes to adapt their function to changing demands and to efficiently express distinctive and modifiable morphomechanical traits in a selective and activity-dependent manner.2,15
Consequently, one-time gene expression profiling of an individual/patient/family-member may be unreliable in determining diagnosis, treatment, or prognosis.2 Serial genetic testing in HCM is necessary, and with new knowledge arises progress in discerning disease causality and pathogenesis, embodied in fast developing human bioinformatics databases maintained by universities/research institutions, such as the Exome Aggregation Consortium (ExAC) database, and the International Genome Sample Resource (IGSR) 1000 Genomes. Actually, the data are already so enormous that it is imperative to develop effective tools, involving artificial intelligence with machine learning and neural networking, which will construct clinical decision-making algorithms capable of sorting out useful information from the databases.
Polygenic HCM Mechanisms Misclassified as Monogenic? An Evolving ControversyThe imperfect yield of HCM genetic testing may reflect an incomplete knowledge of all genes implicated in the disease, expressing themselves in a regulated program. A mutation critical in the early development of HCM may subsequently no longer be active, or mutations invariably active in one stage may no longer be so in subsequent stages in disease evolution.2 Reclassifications of prior “misclassified” pathogenic mutations based on subsequently available genetic and functional information do arise; so also do instances of genotype positive (G+) –phenotype negative (P-) family members, who might well exemplify modifier-gene (polygenic) effects.
Therefore, as recently proposed,15 it appears probable that polygenic phenotype-engendering mechanisms and maladies, such as HCM, may inaccurately be misclassified as monogenic. Precedents are indeed found in other diseases related to mitochondrial dysfunction, such as Barth syndrome exhibiting a dilated cardiomyopathy or Parkinson's disease, originally mislabeled as monogenic/Mendelian. Today, this seems oversimplified, as phenotypic variation within family members implies polygenic interactions and environmental/epigenetic factors as important disease modulators. Their multifaceted actions bring to mind Aristotle's holism, which is embodied in the Epigram, “The whole is more than the sum of its parts.” Wholes are not reducible to their parts because they have emergent properties, arising through de novo interactions among the components. Environmental effects on disease expression can be large, as shown by Parkinson's disease developing in some variant-carriers exposed to pesticides.
There are hundreds of distinct missense MYH7 and MYBPC3 mutations responsible for HCM, and the same missense variants are also found in dilated cardiomyopathy (Table), underscoring the importance of the genomic context (modifier genes that influence the phenotypic expression and severity of pathogenic/causative alleles). Also, both inter- and intrafamilial phenotypic variation for each mutation is explainable, at least in part, by the genetic background: it reworks the expressivity and phenotype variability of causative-variants yielding pleiotropy.15 Nonetheless, observations of intrafamilial variations suggest that, besides mutational heterogeneity, environmental factors must also be involved.
THE REDUCTIVE FALLACY, EPISTASIS, AND ARISTOTLE'S IDEA OF HOLISMWhen explaining complex things in terms of simpler causes, we may commit the reductive fallacy: just because A is associated with B, A is nothing but B; eg, a cathedral is nothing but a heap of stones; a violin sonata is nothing but a sequence of vibrating strings. As Aristotle's holism suggests, the whole is greater than the sum of its parts and contains emergent properties that cannot be discovered through exploration of its parts.
Similarly, epistasis signifies that the effect(s) of a gene is/are dependent on the presence of one or more modifier genes (genetic background/context); additionally, epistatic mutations have different effects in combination than individually.15 Thus, interactions between genes generate larger collections of possible genotypes, and corresponding phenotypes. Epistasis is a major cause of gene multifunctionality. An important aspect of epistasis is that it does not just influence the phenotype, it hides the presence/effect(s) of other gene(s). The total-baldness gene is epistatic to those for blond or red hair. Beyond epistasis, gene-environment interactions further multiply phenotypic variety.2,15,20,21
The Blurred Distinction Between Monogenic and Polygenic/Complex InheritanceGenetic interactions beget interindividual variability in (disease) phenotypes and also blur the distinction between monogenic, and digenic, polygenic or complex inheritance, where the interaction of mutations in 2 or many different genes is required for the expression of various clinical phenotypes.2,15 Genetic diseases most likely exemplify a declining-influence spectrum/progression, extending from a single causative gene affected by modifier genes, through-and-up-to multiple covalent genes with increasingly similar potency. Clearly, modifier and/or covalent genes can modulate the phenotype broadly, rendering genotype-based diagnosis, prognosis and therapy problematical.
Probably caused by diverse combinations of genetic and internal/external environmental (including lifestyle) factors, most of which have not been identified, reduced penetrance and variable expressivity are phenomena typically affecting disorders, such as HCM, historically (mis)labeled as autosomal dominant. So far, a polygenic/multifactorial postulate as to HCM has been elusive; but it is accepted that, contingent upon the particular mutation(s), penetrance depends on age, sex, and conditioning/exercise; all important factors that also affect phenotypic expression in secondary concentric hypertrophy.3,20,21
NONGENETIC FACTORS AFFECTING HCM MUTATION EXPRESSIONToday's personalized cardiology embodies the integration of high-technology genetic/genomic and other laboratory molecular biology studies into clinical practice.2,3,15 The phenotypic diversity typifying HCM evokes a multifactorial nature, with a role for modifier genes and internal/external environmental factors in disease expression (Figure 3). Once the HCM phenotype is developed, however, Pérez-Sánchez et al.1 conclude that sex, hypertension and, most notably, physical activity are not associated with disease severity nor have they a major impact on prognosis in causative mutation-carriers.
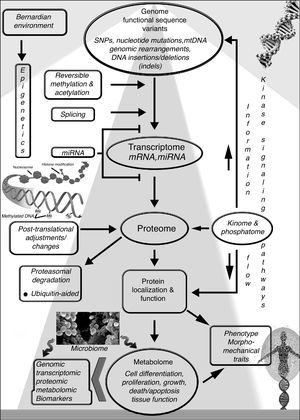
 information flow and signaling pathways variable human gene transcription and expression, while protein levels and function are affected by various post-translational adjustments. Since epigenetic states are reversible, they can be modified by environmental factors, which may contribute to modifiable changes between normal and abnormal phenotypes. Interfering with the activity of factors that modify the chromatin state can possibly affect the expression of unwanted gene-variants. The human organism is effectively a “supraorganism,” a blend of human and microbiome genes and traits, including the metabolome. The relationship between different “-ome/-omics” components, protein levels, post-translational modification, protein localization and activity affecting phenotypic traits, is shown schematically in this general overview. mRNA, messenger RNA; miRNA, microRNA. Reproduced from Pasipoularides,2 with permission of Int J Cardiol and Elsevier.")
The genome integrates intrinsic and environmental signals. The Bernardian environment and epigenetic regulatory factors influence through (bidirectional) information flow and signaling pathways variable human gene transcription and expression, while protein levels and function are affected by various post-translational adjustments. Since epigenetic states are reversible, they can be modified by environmental factors, which may contribute to modifiable changes between normal and abnormal phenotypes. Interfering with the activity of factors that modify the chromatin state can possibly affect the expression of unwanted gene-variants. The human organism is effectively a “supraorganism,” a blend of human and microbiome genes and traits, including the metabolome. The relationship between different “-ome/-omics” components, protein levels, post-translational modification, protein localization and activity affecting phenotypic traits, is shown schematically in this general overview. mRNA, messenger RNA; miRNA, microRNA. Reproduced from Pasipoularides,2 with permission of Int J Cardiol and Elsevier.
A cautionary note, here: the findings of Pérez-Sánchez et al.1 suggest that moderate exercise may not cause problems but modestly-sized clinical trials cannot substantiate that exercise is safe in HMC—that would require randomizing > 2000 patients and following them up for ≈ 3 years. That increased levels of physical activity are not associated with disease severity or prognosis in causative mutation-carriers needs corroboration in larger prospective studies. In this context, the role of exercise-stimulated production, secretion, and expression of cytokines/myokines merits in-depth investigation because their heart benefits might offer an efficient explanation. Indeed, exercise-induced myokines are known to impact many organs.22 In HCM too, exercise-induced myokines might potentially provide beneficial effects,23 by stimulating metabolic pathways, improving glucose uptake, and upgrading fat oxidation.
Indeed, large genotype/epigenotype-phenotype correlative trials can reveal effects of little-known genetic and environmental factors modulating the HCM phenotype, and parallel basic research should clarify the underlying mechanisms. Mechanical stresses, or lack thereof, act as key environmental/epigenetic factors affecting HCM traits.3,20,21 Such factors change the genomic landscape by influencing molecular forces at work; they may include the abnormal absence in the narrowed LV cavity of HCM of the normally strong endo/myocardial shear and “squeeze” forces exerted by large-scale rotatory diastolic right and left ventricular filling flow.24–28
IT IS NOT JUST ABOUT THE GENES, IT IS ABOUT WHAT THE GENES ARE DOINGEnvironmental/epigenetic factors can change the landscape in HCM because it is not only the DNA sequence that influences what is going on. The DNA comprising our genes is incapable of autonomous action, rendering its regulation vital. What a person's genes are induced to do by their contexts—encompassing other genes, besides environmental/epigenetic factors—is just as important as which polymorphic variants are present. An abnormal inherited gene is of no consequence if it remains turned off by epigenetic processes. Epigenetics embraces reversible and sometimes heritable modifications to nucleotides or chromosomes that can change gene expression and chromatin structure without altering the primary DNA nucleotide sequence (Figure 3). Epigenetic modifications include DNA methylation, histone acetylation or methylation, and chromatin architecture.2,15,20,21,25–27 These modifications influence gene regulation and expression, and can implement susceptibility or resistance to various diseases.
Histones, macromolecular proteins associated with DNA packing, can be modified by epigenetic factors through a variety of processes, modifying RNA polymerase access: histone acetylation causes associated DNA segments to become more accessible to RNA polymerase, leading to commencing/enhancing gene transcription/expression. Methyl groups attach directly to CpG dinucleotides in DNA gene promoter segments usually to repress transcription, thus stopping/slowing gene expression.
Epigenetic pathways act at the intersection of genetic and environmental factors (Figure 3). Although it has yet to be understood just how environmental factors alter DNA influencing gene expression throughout life, epigenetics appears to be at the interface of gene-environment (G×E) interactions altering gene expression. This complex issue is pursued energetically in ongoing clinical trials.1 The epigenetic profile of an individual is influenced by both random and environmental factors. Although monozygotic twins are genetically identical, their DNA-methylation patterns become more dissimilar with time. Environmental factors can have both harmful and protective effects through epigenetic processes that may be operative in human disorders including HCM.
Deciphering intricate signaling-chains from mutant protein(s) to clinical phenotype(s), and identification of factors, environmental and epigenetic, which modify the expression of mutations/polymorphic variants, should generate new insights. This makes the work of Pérez-Sánchez et al.1 relevant in seeking a better understanding of the pathogenesis of HCM and in improving preventative and therapeutic interventions.
CONCLUSIONSAlthough much progress has been made in unraveling the pathogenesis of HCM, current understanding is inadequate to predict personalized phenotypes or outcomes. Much remains to be discovered regarding phenotypic penetrance and the possibility of evolving-phenotype prevention or reversal. Disease phenotypes embody interactions between causal genes, modifier genes, and the environment. Advances in functional genomics are becoming increasingly important to cardiology research and practice. Before long, rising genomic/epigenomic sophistication will have a much-anticipated transformative impact on HCM diagnosis, risk stratification, and implementation of therapeutic and preventative measures for patients, mutation-carriers, and families.
FUNDINGResearch support, for work from A. Pasipoularides’ laboratory cited here, was provided by: National Heart, Lung, and Blood Institute, Grant R01 HL 050446; National Science Foundation, Grant CDR 8622201; and North Carolina Supercomputing Center and Cray Research.
CONFLICTS OF INTERESTNone declared.