Keywords
INTRODUCTION
Inherited cardiac conditions are major causes of fatal arrhythmias and sudden cardiac death (SD). Systematic screening of first degree relatives of patients with cardiomyopathies (CMP) and channelopathies (ChP) is recommended. Asymptomatic relatives can be identified, appropriately stratified and effectively treated following a systematic approach. The presence of a family history of SD increases risk in affected relatives with hypertrophic cardiomyopathy.1 In other inherited cardiac diseases the association between family history of sudden cardiac death and risk is yet to be established.2-4
Different triggers, such as exercise and emotional stress, have been associated to SD in type 1 and 2 long QT syndrome (LQTS).5,6 On the contrary, in Brugada syndrome (BrS), similarly to LQT3, SD cases occur typically during increased vagal activity.7,8
Although SD can happen at any age, malignant arrhythmias in CMP and ChP tend to occur more frequently at a particular range of ages.1,9-11 Gender differences in risk have also documented in some of these conditions, such as BrS, LQTS, and arrhythmogenic right ventricular cardiomyopathy (ARVC),9-12 but not in others.1,2
The available information with regards to the context in which SD occurs is scarce and limited to few referral centers. We reviewed SD cases in a large population with different types of CMP and ChP from a dedicated clinic. We aim to report on the circumstances of SD cases in families with inherited diseases.
METHODS
Study Population
One hundred and fifty-two reported SDs (aged, 43 [19], range, 1 month - 82 years) from 103 families with inherited cardiac diseases were included; 36 (23.7%) families had multiple SDs. Reasons for referral of the 103 families with SD cases were: resuscitated cardiac arrest in 10 (6.6%), recent SD case in the family in 11 (7.2%), diagnosis of CMP or ChP in a living relative in 63 (41.5%).
The 103 families were selected from a cohort of 493 families consecutively referred to a cardiac screening clinic during 5 years (February 2003 to May 2008). Three hundred ninety families with diagnosis of CMP or ChP had no SD cases. Total population of 694 living affected individuals (aged, 49 [21]; 439 [63.2%] males) were evaluated during this period of time. Recommended diagnosis criteria were used for hypertrophic cardiomyopathy (HCM),13,14 idiopathic dilated cardiomyopathy (DCM),13,15 ARVC,16 isolated left ventricular non-compaction (iLVNC),17 BrS,18,19 and LQTS.18
SD was defined as unexpected within 1 hour of appearance of symptoms in people under 75 years of age, or in older patients with known CMP or ChP. Successfully resuscitated cardiac arrest was also included as SD equivalent. Patients with non-cardiac or other non-related cardiovascular causes of SD were excluded.
Detailed family trees were taken from interviews with patients and relatives. They were asked about cardiac conditions in the family, particularly about heart failure, transplantation, requirement of pacemakers, other devices or relevant treatments, consanguinity, and about the causes and ages of deaths particularly cardiovascular deaths. Comorbidities, past medical history, cardiac symptoms, diagnosis and relevant therapies of the SD cases prior to the event were recorded. Possible triggers such as exercise, medications, infections and symptoms on the day of the SD were similarly recorded. Autopsies, medical records and ECGs were reviewed when available.
Statistical Analysis
A dedicated database was created for the study. SPSS for PC statistical program (version 11.0) was used for the analysis. Continuous variables are presented as mean (SD). Two-tailed Student t test (continuous variables), c2 test (qualitative variables), or Fisher exact test (qualitative variables with expected counts <5) were used to compare CMP and ChP groups in Table 1 and "Results." Six-hundred ninety-four living patients from the cohort of families and 152 SD cases are included in survival analysis (n=846). Age of death in SD cases and age at last follow-up in affected living patients where used for survival analysis. Kaplan-Meier charts were produced and log rank test was employed for comparisons (Figures 1-3). A P value less than .05 was considered statistically significant.


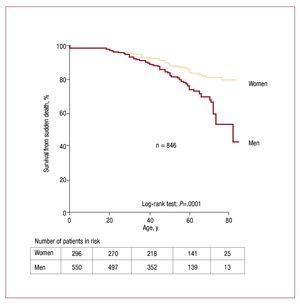
Figure 1. Survival from sudden death in families with inherited diseases and gender.

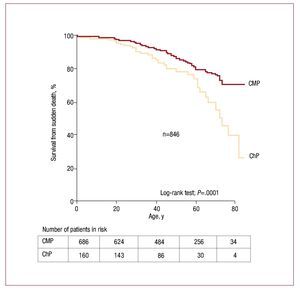
Figure 2. Survival from sudden death (SD) in families with cardiomyopathies (CMP) versus channelopathies (ChP).

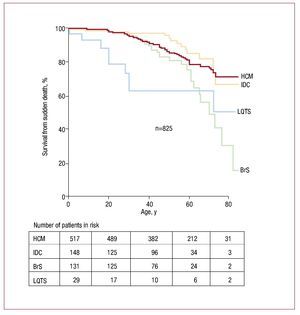
Figure 3. Survival from sudden death in families with different inherited diseases. BrS indicates Brugada syndrome; HCM, hypertrophic cardiomyopathy; IDC, idiopathic dilated cardiomyopathy; LQTS, long QT syndrome.
RESULTS
There were 115 SDs in 77 families with CMP and 37 in 26 families with ChP. Characteristics of the population of SD regarding family diagnosis of CMP and ChP are listed in Table 1. Diagnosis of CMP was known prior to SD in 31.3% versus 13.5% in the ChP group (P=.03). Contrarily in 10.8% of ChP cases diagnosis was reached after resuscitated cardiac arrest versus 1.7% in CMP group (P=.03). Autopsies were not performed or medical records that could give insights into the cause of death were not available in 80 (52.6%) SD cases (48.7% in CMP and 64.9% in ChP group (P=.09)).
Fifty-five (44.4%) patients were on medication at the time of death, 41 (27%) of whom were on cardiac drugs for known cardiac conditions. Only in 1 case was medication thought to be the cause of resuscitated SD in a LQTS patient.
Gender and Age
Male gender was significantly associated to SD cases (111 [73%] vs 41 [27%]; P<.03). Mean age of males and females was similar (43 [19] vs 41 [19]; P=.5), with no differences in the type of condition and gender. Females tended to die slightly more during sleep (12 [29.3%] vs 19 [11.1%]; P=.09) while SD in males occurred more during exercise/stress/ daily activities (59 [53.2%] vs 16 [39%]; P=.1).
When all living patients from the cohort and SD cases were included, survival analysis demonstrated a significantly increased SD mortality in males compared to females (P=.0001) (Figure 1).
Age was significantly related to circumstances of SD. Ten (31.3%) out of the 25 cases of exercise related SD were 1 to 30 years of age (P=.01). There was a similar distribution of circumstances of SD and age in males and females. Age of SD cases from CMP and ChP groups were similar (Table 1).
Age of SD in iLVNC families was slightly but not significantly lower compared to other CMPs (P=.4; mean age, 27 [11] years old for iLVNC, followed by 40 [18] for ARVC, 43 [17] for HCM and 46 [24] for DCM). There was a male gender predominance in all but in iLVNC (75.9% for HCM, 66.7% for ARVC, 57.1% for DCM, and 33.3% for iLVNC). Differences in percentage of male gender between HCM versus non-HCM did not reach significance (P=.08).
SD cases in LQTS families were significantly younger than BrS (25 [20] vs 47 [19]; P=.01) and HCM (P=.03). Males were predominant in both ChP (22, [78.6%] for BrS, and 7 [77.8%] for LQTS).
When all living patients from the cohort and SD cases were included, survival analysis demonstrated a significantly increased SD mortality in ChP compared to CMP group (P=.0001), (Figure 2). SD tend to occur earlier in life in LQTS than in other inherited conditions. Compared to DCM and HCM, survival curve for BrS group is characterized by a more pronounced slope (Figure 3).
There were no differences in age or gender regarding degree relativeness.
Characteristics of Sudden Death
Twenty-seven (17.8%) SDs happened during exercise/stress, 49 (32.2%) related to daily activities, 26 (17.1%) at rest, and 31 (20.4%) during sleep (Table 2). SD tended to occur more during exercise/ stress/daily activities in CMP (63 [61.2%]) compared to ChP (12 [41.4%]; P=.057).

There were 115 SDs (aged, 43 [18]; males, 71.3%) in the CMP group. SD happened during exercise in 33% of ARVC, 21% of DCM, and 16% of HCM, and none in the iLVNC group (P=.5 for HCM vs non-HCM). There was a trend towards a higher percentage of males within exercise/stress/daily activities related SD (49 [67.1%] vs 14 [46.7%]; P=.053), with no difference when exercise was taken in isolation (P=.3) (Table 1).
There were 37 SDs in the ChP group, 28 in BrS, and 9 in LQTS families. Exercise related SD was more frequent, although non-significant, in LQTS compared to BrS (2 [22.2%] vs 3 [10.7%]; P=.6). An important percentage of deaths in the ChP group occurred at rest (34.3%). Of note, none of the SDs in this group were triggered by emotional stress. There were 5 athletes (2 LQTS, 3 BrS), with one SD happening during exercise (from a LQTS family).
Ten (37.0%) cases in the ChP group were on medication at the time of death. Despite a single case of LQTS in whom arrhythmia was related to treatment, insufficient data on type of medication and doses made impossible to draw conclusions in this regard. Relatives interviewed did not recall any remarkable sign of overt febrile episode in the 28 SD from BrS group. Notably, there were 3 SDs occurring within 1 week after surgery (2 orthopedic and 1 abdominal hernia) in the BrS group. There were 2 cases of SD related to delivery (1 obligate carrier from LQTS and 1 case in HCM family).
Sudden Death in Athletes
Twenty (13.2%; aged, 31 [15]; 20 males) SDs occurred in athletes. The majority of athletes died during exercise (10 [50.0%]) compared to the non-athletes (15 [11.4%]; P=.0002); 4 during daily activities, 3 at rest, and 2 during sleep. Athletes were significantly younger (31 [15] vs 44 [19]; P=.005), and more frequently male (20 [100.0%]; P=.004). Thirteen (65%) athletes were from HCM families, (15%) BrS, 2 (10%) LQTS, and 2 (10%) ARVC.
Sudden Infant Death Syndrome
There were 6 reported cases of sudden infant death syndrome and this was not associated to any particular type of cardiac disease in the family (2 HCM, 2 DCM, 1 BrS, and 1 LQTS).
DISCUSSION
Information regarding circumstances of SD is based on results from a few highly selected clinical or pathological centres.5,11,20-23 This is the first series to report context of SD in different inherited cardiac diseases consecutively evaluated in a single center.
Sudden Death Cases and Gender
One of the main and unexpected findings of the study was the global association between sudden death and gender. The dominant trait of inheritance of these conditions does not explain the clearly predominant male gender of the SD cases (75%:25%). Moreover, male gender was more frequent in SD in all different diseases, from CMP to ChP. Male and female SD cases occured in similar circumstances and at a similar distribution of age. It is worth noting, that males were also more common than expected (63%:37%) within living patients seen in the clinic.
This finding is in contrast to the lack of association reported in HCM. Two recent studies with large HCM populations, that included slightly more males (59% and 60%), failed to find any independent association between SD and gender.24,25 In the present study, there were 76% SD within males in HCM families, in contrast to the 59% living HCM males evaluated in the clinic in the last 5 years. The number of male SDs exceeds that expected from the distribution of gender in our HCM population (P=.002).
There are scarce and non-conclusive publications on this aspect on DCM.2,26,27 There was a clear prevalence of males included in 3 studies with large populations of patients (from 70%-78%). Nevertheless, no association has been reported between gender and SD in idiopathic DCM. In keeping with this finding, similar predominance of males was found in the living DCM population (66%) than within the SDs cases (57%) (P=.5) from our study.
Although Brugada syndrome has a dominant trait of inheritance, for uncertain reasons, expression of the disease is male predominant.11 In keeping with this figure, males were predominant both, in living patients with BrS seen in the clinic and among SDs from those families (81% for living BrS and 79% within SD cases; P=.8).
Contrary to our results it has been reported that females with some forms of LQTS have a worse prognosis than males.6,10 Despite a balanced percentage of males-females in living patients with LQTS (8 males; 44%), there were more males within SD cases (8; 80%) (P=.07).
Sudden Death, Exercise, and Athletes
An important percentage of SD cases (18%) in the present study happened during exercise. This percentage clearly exceeds the estimated average daily sport activity in the studied population with only 13% of athletes.
In keeping with other studies the majority of athletes from our population died during exercise (50%).20,28-30 The percentage of non-athletes who died during exercise (11%) is similar to the 9% reported by Corrado et al in a recent paper with a large population.29 A male to female ratio 10:1 of SD cases in athletes has been observed in other series, partly but not wholly explained by the lower number of participating females.28,29 All athletes who died suddenly in the present study were males.
Typical and Frequent Triggers of Sudden Death
Typical triggers have been associated to different diseases.5,7,8,29 In keeping with previous studies exercise related SD was particularly important in ARVC and LQTS,5,22 while BrS had the highest percentage of death at rest/sleep. A higher percentage of exercise related deaths in LQTS has been reported in the literature, up to 68% of LQT1 genetically positive patients, which was unusual (<5%) in LQT2 and LQT3.5 Small numbers and coexistence of different genotypes could account for this intermediate percentage of exercise related SD in the present study. No relevant association between medications and SD could be identified, probably reflecting an uncommon trigger in our population.
In contrast to initial studies,7 no febrile episode has been identified as a possible trigger of SD in BrS families. Up to 45% of arrhythmic events were related to fever in a population of children with BrS.8 Mean age of SD cases in our population was 47 years of age. The lower incidence of pyrexial episodes in adults compared to children could partly explain the difference. Percentage of SD cases in BrS families in our series that were triggered by exercise or happened during normal daily activities was higher than expected.
Globally, 1 in 3 SDs happened during normal daily activities in the absence of any obvious trigger. SD tend to occur more during exercise/daily activities in cardiomyopathies and at rest/sleep in channelopathies.
Value of Autopsy and Family History of Sudden Death
Cardiac conditions such as CMP are often under-diagnosed as causes of SD. A high rate of discrepancy between clinical and autopsy findings has been reported.23 Accuracy of death certificates in SD can be as low as 50%.23 Results from exhaustive cardiac autopsy are essential for appropriate diagnosis and managing of relatives of the deceased. In the present study 23% of SDs were probands that led to screening of first degree relatives.
Percentage of reported negative autopsies in SD cases even in experienced hands can be as high as 20%.23,29,31 Recent publications have reported up to 40% of successful rate for the molecular studies of genes encoding ion channels, ryanodine receptor together with selected desmosome and sarcomeric genes.32,33 Future research on the field should include expert exhaustive pathological study and the help of molecular biology.
The presence of a family history of SD increases risk in affected relatives with hypertrophic cardiomyopathy (HCM).1 Its value for risk stratification is less clear and somewhat controversial in other diseases such as ARVC, DCM, BrS, or LQTS.2,3,4 Nevertheless, patients are managed individually and clinicians often take family history into consideration when evaluating risk, and preventive treatments are indicated particularly if SD cases happen in young first degree relatives or when multiple SDs occur in the family.
Limitations
Only a small percentage of SD cases (27%) with no prior diagnosis had an appropriate postmortem study of the heart, probably a result of the lack of a protocolized program in our region at the time and partly from the assumption that ischaemic heart disease was the most likely cause of SD patients with co-morbidity and elder individuals. Cardiac conditions such as CMP and ChP are often misdiagnosed.23,33 In the present study, young SD cases were more likely to undergo autopsy. Majority of SDs occurred before the diagnosis of the first case of CMP or CHP in the family. In this regard, population of SDs with definitive diagnosis of CMP or ChP was similar in age, gender, and circumstances of death, to that of SDs in relatives with no definitive diagnosis.
Information deriving from the interview of patients and relatives was in some cases scarce and incomplete (context of death was unavailable in 16 [10.5%], and detailed information regarding medications on the day of the event was difficult to obtain). Nevertheless, main messages from the study could be reached, such as: prevalence of SD in inherited cardiac diseases, context of SD in different types of conditions and associations between male gender and SD cases.
CONCLUSIONS
SD is common in inherited cardiac diseases, making up an important number of SD cases. Males are clearly predominant (3:1) among SD cases (irrespective of the type of condition). Most SDs occur during exercise or normal daily activities in cardiomyopathies and during rest or sleep in channelopathies. Percentage of exercise related SD in these conditions is higher than expected.
ABBREVIATIONS
ALVC: arrhythmogenic left ventricular cardiomyopathy
BrS: Brugada syndrome
ChP: channelopathy
CMP: cardiomyopathy
HCM: hypertrophic cardiomyopathy
IDC: idiopathic dilated cardiomyopathy
ILVNC: isolated left ventricular noncompaction
LQTS: long QT syndrome
SD: sudden death
SEE ARTICLE ON PAGES 257-60
This work has been made thanks to a grant from Fundación Española del Corazón and Coca-Cola Spain (2006 and 2007) and has been supported by a partial financial support from la Red de Investigación Cardiovascular RECAVA from Instituto de Salud Carlos III.
Correspondence: Dr. J. R. Gimeno.
Servicio de Cardiología. Hospital Universitario Virgen Arrixaca. Ctra. Murcia-Cartagena, s/n. 30120 El Palmar. Murcia. Spain.
E-mail: jgimeno@secardiologia.es
Received March 30, 2009.
Accepted for publication September 17, 2009.