

Hereditary transthyretin amyloidosis (hATTR) is a disease caused by mutations in the transthyretin gene that frequently shows cardiac involvement due to amyloid deposition in the myocardium. Our objective was to identify cardiac involvement in a Spanish cohort.
MethodsRetrospective multicenter study of patients diagnosed with hATTR with cardiac involvement from Spanish centers. We collected demographic, clinical, and genetic data.
ResultsA total of 181 patients from 26 centers were included (65.2% men, with a median age at diagnosis of 62 years). The most frequent mutations were Val50Met (67.7%) and Val142Ile (12.4%). The main reason for consultation was extracardiac symptoms (69%), mainly neurological. The mean N-terminal pro-B-type natriuretic peptide level was 2145±3586 pg/mL. The most characteristic electrocardiogram findings were a pseudoinfarct pattern (25.9%) and atrioventricular block (25.3%). Mean ventricular thickness was 15.4±4.1mm. Longitudinal strain was reduced in basal segments by 29.4%. Late diffuse subendocardial enhancement was observed in 58.8%. Perugini grade 2 or 3 uptake was observed in 75% of scintigraphy scans. During follow-up, 24.9% of the patients were admitted for heart failure, 34.3% required a pacemaker, and 31.6% required a liver transplant. One third (32.5%) died during follow-up, mainly due to heart failure (28.8%). The presence of non-Val50Met mutations was associated with a worse prognosis.
ConclusionsHATTR cardiac amyloidosis in Spain shows heterogeneous genetic and clinical involvement. The prognosis is poor, mainly due to cardiac complications. Consequently early diagnosis and treatment are vital.
Keywords
Cardiac amyloidosis (CA) is a deposition disease caused by extracellular accumulation of abnormal fibers in the heart.1 The amyloid light-chain (AL) form has traditionally been considered more common, but increased study in recent years of amyloidosis secondary to abnormal transthyretin, whether in its hereditary form due to TTR mutations (hATTR) or in its natural or wild-type (wtATTR) form, has led to a greater understanding and to advances in imaging techniques, such that wtATTR is now considered far more prevalent than AL.2,3
hATTR is an autosomal dominant disease caused by transthyretin gene mutations, with more than 120 mutations described to date. The most common is the Val50Met mutation. Identifying patients with amyloidosis due to a genetic defect is enormously important, as it affects the approach to treatment and is greatly relevant for relatives, due to the type of inheritance pattern. Although it is very rare worldwide (estimated prevalence<1/100 000 inhabitants), there are reports of some endemic foci, such as Portugal, Sweden, Brazil, Japan, and 2 Spanish regions (Mallorca and Huelva). Portugal has the highest prevalence globally, with an estimated rate of 1 per 538 inhabitants, whereas Mallorca has a prevalence of around 1 per 3500, the fifth highest level worldwide.4
In hATTR, CA is one of the main causes of morbidity and mortality.5 Approved treatments for hATTR include liver transplant, tafamidis, patisiran, and inotersen. Several clinical trials have already shown that these drugs are beneficial for the treatment of CA.6–8
In Spain, there are few published data on hATTR with cardiac involvement.5,9 Early diagnosis of this disease is essential to identify patients who may benefit from the new treatments.
The aim of our study was to describe the demographic, clinical, and genetic characteristics of patients with hATTR with cardiac involvement in Spain from the whole country, including both endemic foci and nonendemic areas.
METHODSThe design consisted of a descriptive, retrospective, multicenter study of patients diagnosed with hATTR with cardiac involvement. Most of the heart failure units and familial heart disease units in Spain were invited to participate in the study. The study was approved by the ethics committee of the autonomous community of the Balearic Islands, and informed consent was obtained from all patients included.
Patients were required to have a positive genetic study for some mutation in the transthyretin gene. CA was defined as the presence of typical signs in the echocardiogram or cardiac magnetic resonance imaging+histologic confirmation (amyloid present in subcutaneous fat, rectum, salivary gland, myocardium, or another location) or scintigraphy scan with grade 2 or 3 uptake.10 In geographic areas where the Val50Met mutation is endemic, the presence of the mutation+consistent symptoms+typical findings on echocardiography or cardiac magnetic resonance imaging (≥ 13mm in the absence of hypertension or aortic valve disease) was considered sufficient, because of the low sensitivity of scintigraphy in this particular subgroup.11
Each participating hospital reviewed the medical histories of patients with hATTR with cardiac involvement. Data were collected on general demographics, comorbidities, symptoms, genetics, biomarkers, electrocardiogram, echocardiogram, Holter test, cardiac magnetic resonance imaging, myocardial scintigraphy scan with bisphosphonates, biopsy, treatments, complications, and clinical progress.
Statistical analysisA descriptive study was performed by calculating the frequencies of qualitative variables, as well as the median [interquartile range] for quantitative variables. The Kolmogorov-Smirnov test was used to determine Gaussian distribution. Categorical variables are expressed as percentages. Normal quantitative variables were compared using the Mann-Whitney U-test nonparametric test, and qualitative variables by the chi-square test; 95% confidence intervals (95%CI) were calculated for variables with a statistically significant difference. In the case of the most prevalent mutations (Val50Met and Val142Ile), a comparative analysis was carried out between groups. A survival analysis was performed from the start of follow-up by the Kaplan-Meier method, both for all patients and for the most prevalent mutations. Statistical significance was set at a P value <.05 in all cases. Excel 2007 and SPSS v15.0 were used for the statistical analysis.
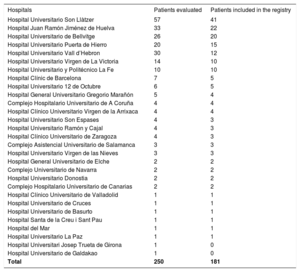
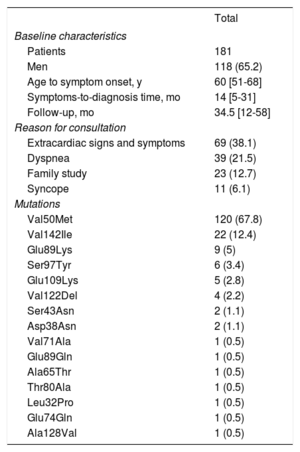
RESULTSInformation was obtained from 250 patients; however, 69 were excluded because they had no evidence of cardiac involvement or were duplicates (patients followed up or studied at 2 participating hospitals). The total number of hospitals contacted was 53, but 27 had no cases of hATTR. The final study included 181 patients from 26 hospitals in Spain who had been diagnosed with hATTR with cardiac involvement. Table 1 lists the distribution according to hospital. Most patients were men (65.2%), the median age at diagnosis was 62 years, and symptom onset occurred at age 58.8±14 years. The time between symptom onset and hATTR diagnosis was 14 months. The median follow-up from the first assessment was 34.5 months. The main presenting complaint was extracardiac symptoms (38.1%), followed by dyspnea (21.5%) (table 2).
Distribution by hospital
| Hospitals | Patients evaluated | Patients included in the registry |
|---|---|---|
| Hospital Universitario Son Llàtzer | 57 | 41 |
| Hospital Juan Ramón Jiménez de Huelva | 33 | 22 |
| Hospital Universitario de Bellvitge | 26 | 20 |
| Hospital Universitario Puerta de Hierro | 20 | 15 |
| Hospital Universitario Vall d’Hebron | 30 | 12 |
| Hospital Universitario Virgen de La Victoria | 14 | 10 |
| Hospital Universitario y Politécnico La Fe | 10 | 10 |
| Hospital Clínic de Barcelona | 7 | 5 |
| Hospital Universitario 12 de Octubre | 6 | 5 |
| Hospital General Universitario Gregorio Marañón | 5 | 4 |
| Complejo Hospitalario Universitario de A Coruña | 4 | 4 |
| Hospital Clínico Universitario Virgen de la Arrixaca | 4 | 4 |
| Hospital Universitario Son Espases | 4 | 3 |
| Hospital Universitario Ramón y Cajal | 4 | 3 |
| Hospital Clínico Universitario de Zaragoza | 4 | 3 |
| Complejo Asistencial Universitario de Salamanca | 3 | 3 |
| Hospital Universitario Virgen de las Nieves | 3 | 3 |
| Hospital General Universitario de Elche | 2 | 2 |
| Complejo Universitario de Navarra | 2 | 2 |
| Hospital Universitario Donostia | 2 | 2 |
| Complejo Hospitalario Universitario de Canarias | 2 | 2 |
| Hospital Clínico Universitario de Valladolid | 1 | 1 |
| Hospital Universitario de Cruces | 1 | 1 |
| Hospital Universitario de Basurto | 1 | 1 |
| Hospital Santa de la Creu i Sant Pau | 1 | 1 |
| Hospital del Mar | 1 | 1 |
| Hospital Universitario La Paz | 1 | 1 |
| Hospital Universitari Josep Trueta de Girona | 1 | 0 |
| Hospital Universitario de Galdakao | 1 | 0 |
| Total | 250 | 181 |
Patients’ clinical and genetic profile
| Total | |
|---|---|
| Baseline characteristics | |
| Patients | 181 |
| Men | 118 (65.2) |
| Age to symptom onset, y | 60 [51-68] |
| Symptoms-to-diagnosis time, mo | 14 [5-31] |
| Follow-up, mo | 34.5 [12-58] |
| Reason for consultation | |
| Extracardiac signs and symptoms | 69 (38.1) |
| Dyspnea | 39 (21.5) |
| Family study | 23 (12.7) |
| Syncope | 11 (6.1) |
| Mutations | |
| Val50Met | 120 (67.8) |
| Val142Ile | 22 (12.4) |
| Glu89Lys | 9 (5) |
| Ser97Tyr | 6 (3.4) |
| Glu109Lys | 5 (2.8) |
| Val122Del | 4 (2.2) |
| Ser43Asn | 2 (1.1) |
| Asp38Asn | 2 (1.1) |
| Val71Ala | 1 (0.5) |
| Glu89Gln | 1 (0.5) |
| Ala65Thr | 1 (0.5) |
| Thr80Ala | 1 (0.5) |
| Leu32Pro | 1 (0.5) |
| Glu74Gln | 1 (0.5) |
| Ala128Val | 1 (0.5) |
Data are expressed as No. (%) or median [interquartile range].
Missense mutations were the most common (97.7%). A total of 15 different mutations were reported, with Val50Met observed most often (n=120; 67.8%), followed by Val142Ile (n=22; 12.4%). Among the latter, only 1 patient was of black race (table 3). Even when disregarding the 2 endemic foci of Val50Met, this mutation remained the most common (n=60; 51.2%) in the rest of Spain (table 2).
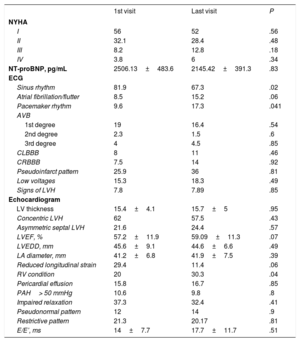
Baseline and follow-up clinical and examination characteristics
| 1st visit | Last visit | P | |
|---|---|---|---|
| NYHA | |||
| I | 56 | 52 | .56 |
| II | 32.1 | 28.4 | .48 |
| III | 8.2 | 12.8 | .18 |
| IV | 3.8 | 6 | .34 |
| NT-proBNP, pg/mL | 2506.13±483.6 | 2145.42±391.3 | .83 |
| ECG | |||
| Sinus rhythm | 81.9 | 67.3 | .02 |
| Atrial fibrillation/flutter | 8.5 | 15.2 | .06 |
| Pacemaker rhythm | 9.6 | 17.3 | .041 |
| AVB | |||
| 1st degree | 19 | 16.4 | .54 |
| 2nd degree | 2.3 | 1.5 | .6 |
| 3rd degree | 4 | 4.5 | .85 |
| CLBBB | 8 | 11 | .46 |
| CRBBB | 7.5 | 14 | .92 |
| Pseudoinfarct pattern | 25.9 | 36 | .81 |
| Low voltages | 15.3 | 18.3 | .49 |
| Signs of LVH | 7.8 | 7.89 | .85 |
| Echocardiogram | |||
| LV thickness | 15.4±4.1 | 15.7±5 | .95 |
| Concentric LVH | 62 | 57.5 | .43 |
| Asymmetric septal LVH | 21.6 | 24.4 | .57 |
| LVEF, % | 57.2±11.9 | 59.09±11.3 | .07 |
| LVEDD, mm | 45.6±9.1 | 44.6±6.6 | .49 |
| LA diameter, mm | 41.2±6.8 | 41.9±7.5 | .39 |
| Reduced longitudinal strain | 29.4 | 11.4 | .06 |
| RV condition | 20 | 30.3 | .04 |
| Pericardial effusion | 15.8 | 16.7 | .85 |
| PAH> 50 mmHg | 10.6 | 9.8 | .8 |
| Impaired relaxation | 37.3 | 32.4 | .41 |
| Pseudonormal pattern | 12 | 14 | .9 |
| Restrictive pattern | 21.3 | 20.17 | .81 |
| E/E’, ms | 14±7.7 | 17.7±11.7 | .51 |
AVB, atrioventricular block; CLBBB, complete left bundle branch block; CRBBB, complete right bundle-branch block; ECG, electrocardiogram; LA, left atrium; LV, left ventricle; LVEDD, left ventricular end-diastolic diameter; LVEF, left ventricular ejection fraction; LVH, left ventricular hypertrophy; NT-proBNP, N-terminal fraction of pro-brain natriuretic peptide; NYHA, New York Heart Association functional class; PAH, pulmonary arterial hypertension; RV, right ventricle.
The data are expressed as mean ± standard deviation or percentage.
At the first visit, most patients were in New York Heart Association functional class I or II, and up to 12% were in functional class III or IV. The mean N-terminal pro-B type natriuretic peptide (NT-proBNP) level was 2506±483 pg/mL. There were no significant differences in functional class or NT-proBNP levels during follow-up. Electrocardiography at the first visit revealed some kind of atrioventricular block in 25.3%, atrial fibrillation or flutter in 8.5%, and pacemaker rhythm in 9.6%. Pseudoinfarct pattern was present in 25.9% of electrocardiograms, and 15.3% showed low voltage. A total of 7.8% of patients had signs of left ventricular hypertrophy. At the last visit, a higher percentage had pacemaker rhythm (17.3% vs 9.6%; P=.041) and pseudoinfarct pattern (25.9% vs 36%; P=.81). On echocardiography, mean ventricular thickness was 15.4±4.1mm, and ventricular hypertrophy was concentric in 62%. Left ventricular ejection fraction was 57.2%±11.9%. The mean anteroposterior diameter of the left atrium was 41.2±6.8mm. In 29.4% of patients, longitudinal strain was reduced in basal segments compared with apical segments. Pericardial effusion was observed in 15.8% of patients, but most (87.9%) cases were mild. A total of 21.3% had a restrictive diastolic pattern. At the last visit, right ventricle involvement was seen more often (20% vs 30.3%; P=.04) (table 3).
In patients who underwent cardiac magnetic resonance imaging (n=71), a diffuse subendocardial pattern was observed in 52.7% and a focal pattern in 16.3%. T1 mapping was rarely used (10 patients), and native T1 was prolonged in half these patients.
Cardiac scintigraphy with bisphosphonates (n=80) revealed Perugini grade 2 to 3 uptake in 75% of cases. Grade 1 uptake was seen in 18.75% and grade 0 in 6.25%.
Biopsies were taken from 93 (52.5%) patients. Among the positive biopsies, 23 (27.4%) were of subcutaneous fat, 19 (22.6%) of myocardium, 15 (17.9%) of rectal mucosa, and 15 (17.9%) of the sural nerve.
Extracardiac involvement was common: neurologic in 68%, gastrointestinal in 35.4%, and renal in 16.4%. A total of 25.8% had a history of carpal tunnel syndrome, and 42.5% had a history of signs of dysautonomia. Neuropathy and gastrointestinal involvement were more common in patients with the Val50Met mutation (80% vs 27.2%; P <.001) than with Val142Ile (39.1% vs 4.5%, P=.006), whereas carpal tunnel syndrome was more common with the Val142Ile mutation than with Val50Met (54.5% vs 11.6%; P <.001) (table 4).
Extracardiac condition at the first assessment
| Total (n=181) | Val50Met (n=120) | Val142Ile (n=22) | P | |
|---|---|---|---|---|
| Carpal tunnel syndrome | 46 (25.8) | 14 (11.6) | 12 (54.5) | <.001 |
| Neuropathy | 123 (68) | 96 (80) | 6 (27.2) | <.001 |
| Dysautonomia | 74 (42.5) | 48 (41.6) | 6 (27.2) | .25 |
| Digestive condition | 64 (35.4) | 51 (39.1) | 1 (4.5) | .006 |
| Renal condition | 29 (16.4) | 20 (17) | 3 (13.6) | .72 |
Data are expressed as No. (%).
A pacemaker was implanted in a third (34.3%) of patients. In half (49.2%) these patients, the pacemaker was indicated due to complete atrioventricular block, 23.1%, as prophylaxis before liver transplant, and 18.5% due to sinus node dysfunction. An implantable defibrillator was not prescribed often, and was only implanted in 4% of patients.
Baseline medical treatment consisted of diuretics in 41.5% of patients, angiotensin-converting enzyme inhibitors in 30.2%, beta-blockers in 17.5%, aldosterone antagonists in 11.9%, and anticoagulants in 20.9%. The specific treatment for hATTR was tafamidis in 25.4% of patients, diflunisal in 24%, liver transplant in 31.6%, and heart transplant in 4.5%.
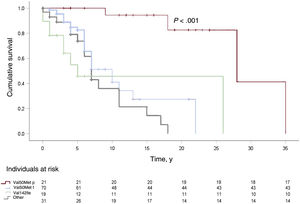
During a median follow-up of 34.5 months, 60.2% required hospitalization mainly due to a cardiac (59.6%) or neurologic (17.4%) cause: heart failure in most (67%) patients in the first group and stroke in most (57.8%) of those in the second group. A third (32.5%) of patients died, mainly due to heart failure (28.8%), sudden cardiac death (11.5%), or a neurologic or renal cause (17.3%). Survival analyses are shown in figure 1 and figure 2.
 and from symptom onset to the combined endpoint of death or heart transplant (B). Median of 8 and 11 years, respectively.")
, Val50Met (late onset), Val142Ile, and all other mutations, from symptom onset to the combined endpoint of death or heart transplant. Median of 28, 10, 5, and 7 years, respectively.")
Survival curves for patients with hereditary transthyretin amyloidosis according to genotype: Val50Met (early onset), Val50Met (late onset), Val142Ile, and all other mutations, from symptom onset to the combined endpoint of death or heart transplant. Median of 28, 10, 5, and 7 years, respectively.
This is the first study to provide a broad and representative description of the main characteristics of patients with hATTR in Spain. To date, there are limited published data, reported by only 2 Spanish hospitals.5,9
Diagnosis of cardiac involvement in hATTR has improved in recent years with noninvasive methods based on bone scintigraphy and cardiac magnetic resonance imaging.12 However, the advent of specific treatments has raised interest in early diagnosis.
In general, the most common mutation in Spain is by far Val50Met (67.8%), not only in the 2 endemic foci of this mutation (Mallorca and Huelva), but also in the rest of Spain, although at a lower percentage (51.2%). There is considerable variability in the mutations observed in Spain, with a total of 15 variants (table 2). Val142Ile is the most common in a large part of Europe and the United States, but is the second most common in Spain (18.8%).
Historically, the Val50Met mutation has been considered almost purely neurologic.4 Our study shows that heart involvement is common in these patients. The THAOS registry13 reports that the cardiac phenotype is mainly due to cardiac mutations (Val142Ile, Leu131Met, and Ile88Leu) in Europe and Val142Ile in the United States,14 although cardiac involvement is also seen often with Val50Met.
The presentation of hATTR is highly heterogeneous, with most patients exhibiting concentric left ventricular hypertrophy and atrial growth. This study revealed some of the keys for suspecting the condition in patients: heart failure with preserved ejection fraction, pseudoinfarct pattern, low voltage or conduction disorders, right ventricular impairment, or mild pericardial effusion or longitudinal strain gradient between the base and the apex, among others. Clinicians should pay attention to extracardiac conditions that may be present in most patients, particularly neuropathy and gastrointestinal conditions in patients with Val50Met and carpal tunnel syndrome in patients with Val142Ile. This mixed phenotype was already seen to be common in the European cohort of the THAOS registry,13 although our study shows that cardiac involvement is still higher than the level reported in publications and underscores the importance of a multidisciplinary assessment of patients with hATTR by various specialties.
Despite the increase in the number of diagnoses of hATTR with heart involvement, improvements can still be made, as the time from the symptom onset to diagnosis is 1.9±2.3 years. Similar delays are described in other studies, with delays of up to 5 years in patients with no family history.15
Our study found 5 patients (6% of patients who had undergone scintigraphy) with Perugini grade 0 uptake. The current protocol for CA diagnosis indicates that grade 2 to 3 uptake is necessary in the scintigraphy scan; however, several published studies, particularly in patients with Val50Met, early onset, and predominantly neurologic symptoms, report that this test has low sensitivity (as low as 41%).11,16 In our series, all these patients had neurologic involvement, 3 of them had the Val50Met mutation, and 2 had the Ser97Tyr mutation with neurologic involvement. In these last 2 patients, cardiac magnetic resonance revealed diffuse subendocardiac gadolinium late-enhancement consistent with a diagnosis of CA and, in the 3 patients with the Val50Met mutation that had no degree of uptake, 2 had ventricular hypertrophy not explicable by other causes plus conduction disorders (first-degree atrioventricular block and second degree Mobitz I, respectively) and 1 had syncope due to complete atrioventricular block and a pacemaker at age less than 60 years.
Common treatments in our series were beta-blockers, angiotensin-converting enzyme inhibitors, and other standard-of-care treatments for heart failure, but require caution, as they are usually poorly tolerated and can be harmful, particularly in the case of beta-blockers.17 In our series, only 1 of every 5 patients was receiving anticoagulant therapy. The high prevalence of intracardiac thrombi and strokes in CA tends to increase mortality and, therefore, emboligenic risk stratification should be undertaken carefully and early anticoagulation therapy is usually recommended.18 It is believed that some centers do not use the CHA2DS2-VASc criteria correctly. In the present series, the prevalence of atrial fibrillation/atrial flutter was only 8.5%, rising to 15.2% during follow-up, and the left atrium was 41.2±6.8mm at baseline and 41.9±7.5mm after follow-up. It is thought that these percentages, along with the other factors already mentioned, influence physicians’ decision not to prescribe anticoagulants to more patients.
Mortality was high in our series, most often due to cardiac causes, such as heart failure and sudden cardiac death. Nevertheless, defibrillator implantation is rare, probably because extracardiac involvement negatively affects patient prognosis in many cases or because the heart disease is already well advanced. Furthermore, the benefit of defibrillators in primary prevention has not been clearly demonstrated.19 The high rate of decompensation due to heart failure and heart transplant and the high mortality suggests that the disease is diagnosed in an advanced stage and that early diagnosis of this condition should be improved to use the treatments shown to be beneficial in early stages.
The survival analysis revealed that the various Val50Met mutations are usually associated with poorer prognosis and that, moreover, the Val50Met mutation with early onset (< 50 years) (more neurologic phenotype) has a better prognosis than the other mutations (late-onset Val50Met, Val142Ile, and others) (more cardiologic phenotype). This is in keeping with other articles showing that mutations of predominantly cardiac phenotype have a worse prognosis.20 Likewise, the survival analyses suggest that the prognosis of these patients is improved if there are fewer years between symptom onset and diagnosis.
Last, more than 80% of patients had received some specific treatment for hATTR, either liver transplant, tafamidis, or diflunisal, likely yielding a significant beneficial impact on their clinical course in many patients. A fourth of the total series received treatment with tafamidis, which was prescribed with a neurologic indication (20mg/d) rather than to treat heart disease, as the data collected in this study were prior to the publication of the ATTR-ACT study,6 which used a much higher dose. Additionally, the study data were collected between 2018 and 2019, and at that time the Spanish Agency of Medicinal Products had not yet approved the TTR inhibitors (patisiran and inotersen).
CONCLUSIONShATTR often manifests with cardiac involvement. This study describes the main characteristics of this disease in the Spanish population. There is a broad range of mutations, although the most common is Val50Met. Extracardiac involvement is frequent, particularly neuropathy and carpal tunnel syndrome. Now that noninvasive methods are available to evaluate cardiac conditions, clear information on this heterogeneous disease will aid in early diagnosis and treatment.
FUNDINGThis study received funding from a research grant of the Heart Failure Section of the Spanish Society of Cardiology in 2018.
AUTHORS’ CONTRIBUTIONSStudy design and database creation: Tomás Ripoll-Vera, Jorge Álvarez Rubio. Patient selection and inclusion of variables in the database: Jorge Álvarez Rubio, Ana José Manovel Sánchez, José González-Costello, Pablo García-Pavía, Javier Limeres Freire, José Manuel García-Pinilla, Esther Zorio Grima, Ana García-Álvarez, María Valverde Gómez, M. Ángeles Espinosa Castro, Gonzalo Barge-Caballero, Juan Ramón Gimeno Blanes, María Teresa Bosch Rovira, Luis Miguel Rincón Díaz, Miguel Ángel Aibar Arregui, María Gallego-Delgado, Juan Jiménez-Jáimez, Marina Martínez Moreno, Mayte Basurte, Xabier Arana Achaga, Idaira Famara Hernández Baldomero. Results assessment: Jorge Álvarez Rubio, Tomás Ripoll-Vera. Writing of manuscript: Jorge Álvarez Rubio, Tomás Ripoll-Vera. Critical review of the manuscript: José González-Costello, Ana José Manovel Sánchez, Pablo García-Pavía, Javier Limeres Freire, José Manuel García-Pinilla, Esther Zorio Grima, Ana García-Álvarez, María Valverde Gómez, M. Ángeles Espinosa Castro, Gonzalo Barge-Caballero, Juan Ramón Gimeno Blanes, María Teresa Bosch Rovira, Luis Miguel Rincón Díaz, Miguel Ángel Aibar Arregui, María Gallego-Delgado, Juan Jiménez-Jáimez, Marina Martínez Moreno, Mayte Basurte, Xabier Arana Achaga, Idaira Famara Hernández Baldomero.
CONFLICTS OF INTERESTJ. González-Costello has received fees for lectures at congresses and courses for Pfizer and Alnylam, has provided consulting services to Pfizer and Alnylam, and is the current chair of the Heart Failure Association of the Spanish Society of Cardiology. E. Zorio Grima has received fees for lectures at congresses and courses for Alnylam. A. García-Álvarez has received fees for lectures at congresses and courses for Novartis, Rovi, and AstraZeneca and currently holds a patent. G. Barge-Caballero has received a research grant payment, has received fees for lectures at congresses and courses for Pfizer, AstraZeneca, Novartis, and Servier and has provided consulting services to Pfizer. M. Basurte has received consulting fees from Pfizer. T. Ripoll-Vera obtained a research grant payment from the Heart Failure Association of the Spanish Society of Cardiology. All other authors declare no conflict of interests.
- –
hATTR is a multisystemic disease with a higher prevalence than previously thought and is clearly underdiagnosed.
- –
Diagnosis is late due to poor understanding among the medical community and variable disease expression.
- –
Considerable advances have been made in recent years in noninvasive diagnosis and in new molecules to improve the prognosis of the disease.
- –
The overall genotypic and phenotypic profile of hATTR in Spain has been unknown until now.
- –
The clinical presentation and characteristics of patients with hATTR in Spain is heterogeneous and differs substantially by genotype; cardiac involvement is very common.
- –
In Spain, hATTR is increasingly diagnosed by noninvasive methods.
To Catalina Meliá Mesquida and Yolanda Gómez Pérez for their work in patient coordination and research management in the Familial Heart Disease Unit, and to the ATTR Multidisciplinary Group of the Hospital Universitario of Son Llàtzer.
Ainara Lozano Bahamonde (Hospital Universitario Basurto), Coloma Tirón (Hospital Universitari Dr. Josep Trueta, Girona), José Onaindia Gandarias and Ángela Cacicedo Fernández de Bobadilla (H. Galdakao, Bizkaia), Pablo Elpidio García Granja and Javier López Díaz (Hospital Clínico Universitario Valladolid), Amaria Núñez Íñiguez (Hospital Universitario Cruces, Bilbao), Mercedes Rivas-Lasarte (Hospital Santa Creu i Sant Pau, Barcelona), Sonia Ruiz Bustillo (Hospital del Mar, Barcelona), Juan Caro (Hospital Universitario La Paz, Madrid), Marta Padilla-Sainz, Carlos Casasnovas, Carmen Baliellas, Laura Lladó, Emma González-Vilatarsana, and Carles Díez-López (Hospital Universitari de Bellvitge, Barcelona), José F. Rodríguez-Palomares (Hospital Universitario Vall d’Hebron, Barcelona), Ainhoa Robles-Mezcua and Arancha Díaz-Expósito (Hospital Universitario Virgen de la Victoria, Málaga), Javier Navarrete-Navarro (Hospital Universitario y Politécnico La Fe, Valencia), José T. Ortiz-Pérez (Hospital Clínic-IDIBAPS, Barcelona), Julián Palomino-Doza and Rafael Salguero-Bodes (Hospital Universitario 12 de Octubre, Madrid; CIBERCV), Irene Méndez Fernández (Hospital Gregorio Marañón, Madrid), Roberto Barriales-Villa and José M. Larrañaga-Moreira (Complexo Hospitalario Universitario de A Coruña), Elena Fortuny Frau and Jaume Pons Llinares (Hospital Universitario Son Espases, Palma de Mallorca), Pablo Revilla Martí (Hospital Clínico Universitario de Zaragoza, IIS-A, Zaragoza), Eduardo Villacorta Argüelles (Complejo Asistencial Universitario de Salamanca, Salamanca), Rosa Macías Ruiz (Hospital Universitario Virgen de las Nieves, Granada), Irene Rilo Miranda and Itziar Solla Ruiz (Hospital Universitario Donostia, San Sebastián), Antonio Lara Padrón and Francisco Bosa Ojeda (Complejo Hospitalario Universitario de Canarias), Paula Morlanes Gracia and Guido Antoniutti (Hospital Universitario Son Llàtzer, Palma de Mallorca).