
Mutations in the troponin T gene (TTNT2) have been associated in small studies with the development of hypertrophic cardiomyopathy characterized by a high risk of sudden death and mild hypertrophy. We describe the clinical course of patients carrying mutations in this gene.
MethodsWe analyzed the clinical characteristics and prognosis of patients with mutations in the TNNT2 gene who were seen in an inherited cardiac disease unit.
ResultsOf 180 families with genetically studied cardiomyopathies, 21 families (11.7%) were identified as having mutations in TNNT2: 10 families had Arg92Gln, 5 had Arg286His, 3 had Arg278Cys, 1 had Arg92Trp, 1 had Arg94His, and 1 had Ile221Thr. Thirty-three additional genetic carriers were identified through family assessment. The study included 54 genetic carriers: 56% were male, and the mean average age was 41 ± 17 years. There were 33 cases of hypertrophic cardiomyopathy, 9 of dilated cardiomyopathy, and 1 of noncompaction cardiomyopathy, and maximal myocardial thickness was 18.5 ± 6mm. Ventricular dysfunction was present in 30% of individuals and a history of sudden death in 62%. During follow-up, 4 patients died and 14 (33%) received a defibrillator (8 probands, 6 relatives). Mean survival was 54 years. Carriers of Arg92Gln had early disease development, high penetrance, a high risk of sudden death, a high rate of defibrillator implantation, and a high frequency of mixed phenotype.
ConclusionsMutations in the TNNT2 gene were more common in this series than in previous studies. The clinical and prognostic profiles depended on the mutation present. Carriers of the Arg92Gln mutation developed hypertrophic or dilated cardiomyopathy and had a significantly worse prognosis than those with other mutations in TNNT2 or other sarcomeric genes.
Keywords
Hypertrophic cardiomyopathy (HCM) is an inherited autosomal-dominant disease with a heterogeneous clinical presentation and natural history,1 and is a frequent cause of sudden cardiac death (SCD) in young people2–4; it is associated with mutations in genes coding for sarcomere proteins.5–7 In the literature, debate surrounds the genotype-phenotype correlation of individual mutations,7,8 concerning establishing a prognosis according to the mutation present, which could help stratify the disease and allow appropriate genetic counselling to families. Mutations in the troponin T gene (TNNT2) were described years ago in several publications with few families, and researchers postulated a high prevalence of SCD in young carriers5,6,9,10 who, in addition, had a phenotype of mild left ventricular hypertrophy.6,11
This study aimed to describe the clinical course of a series of patients and relatives—a relatively large series considering the low prevalence of the disease—who were carriers of mutations in TNNT2 and to expand existing knowledge on their prognosis.
METHODSThe cohort was made up of apparently unrelated probands with cardiomyopathy, most with an HCM phenotype. They were assessed in a familial heart disease clinic in Hospital Son Llàtzer (Palma de Mallorca, Balearic Islands, Spain) over a 7-year period, undergoing genetic study of mutations in the TNNT2 gene (and 4 other sarcomeric genes: MYBPC3, MYH7, TNNI3, and TPM1, as well as lamin A/C if the proband had a dilated cardiomyopathy [DCM] phenotype). All relatives of carriers were offered clinical and genetic assessment.
Hypertrophic cardiomyopathy was diagnosed when the maximal myocardial thickness (MMT) was ≥ 15mm in at least 1 segment in the absence of other diseases to explain the hypertrophy.12,13 In probands with SCD as the first clinical manifestation, the diagnosis of HCM was confirmed at autopsy whenever possible. Relatives were considered affected when they met the HCM familial criteria (≥ 13mm).14
All patients and relatives underwent electrocardiography, echocardiography, stress testing, and 24hours Holter monitoring, as per the methods described,15 as well as cardiac magnetic resonance whenever possible.
The main risk factors for SCD were defined as a family history of SCD, syncopal episode of arrhythmic origin or unknown etiology, nonsustained ventricular tachycardia ≥ 120 bpm, MMT ≥ 30mm and abnormal blood pressure response to exercise (in those younger than 40 years).16
The TNNT2 gene was sequenced with Sanger sequencing or next generation sequencing (NGS). Of 21 probands, 19 were studied using Sanger (the 5 main sarcomeric genes were sequenced: MYBPC3, MYH7, TNNT2, TNNI3, and TPM1) and 2 were studied with NGS (in one patient, 12 genes: the 5 sarcomeric genes plus ACTC1, GLA, MYL2, MYL3, PRKAG2, PTPN11, and TNNC1, and in the other patient, 27 genes: the previous 12 plus CASQ2, DMD, DTNA, FKBP1A, KCNH2, LDB3, LMNA, MIB1, MYH6, NOTCH1, PLN, RYR2, SCN5A, TAZ, and TTN, the latter combination because of the noncompaction phenotype). A change in the amino acid sequence compared with the reference sequence was considered a pathogenic mutation when it met the following criteria: it segregated in the affected family members, it was not present in 200 chromosomes from healthy unrelated individuals, it had not yet been identified in populations of thousands of individuals from different ethnic groups included in the 5000 Genomes Project (Exome Variant Server), the 1000 Genomes Project, or dbSNP (Short Genetic Variations database), and it affected a residue that is phylogenetically conserved between troponin T species and isoforms. An allelic variant was considered rare when segregation could not be demonstrated and the variant was not present in controls, and polymorphic when it was not associated with the disease and it was present in controls. The previously described variants were reviewed to assess their pathogenicity, and new mutations were studied with in silico tools.
Informed consent for DNA extraction was obtained from each individual. The study adhered to the principles of the Declaration of Helsinki, the Council of Europe Convention on Human Rights and Biomedicine, and the UNESCO Universal Declaration on the Human Genome and Human Rights.
Statistical analysis was performed using the SPSS application (v.15.0, SPSS Inc.; Chicago, Illinois, United States). Data with normal distribution are expressed as means (95% confidence interval). The differences between the means were compared using an unpaired 2-tailed Student t test. Categorical data were compared using a chi-square test. Continuous data with abnormal distribution were analyzed with a Mann-Whitney U test. The predefined outcomes for survival analysis were as follows: SCD, first appropriate shock from implantable cardioverter-defibrillator (ICD), death due to heart failure, cardiac transplant, and other cardiovascular death. The cumulative probability for the occurrence of an event was calculated using the Kaplan-Meier method. A comparison was performed with previously published data. Survival analysis from birth was also performed for individual mutations. A probability value of P < .05 was considered statistically significant.
RESULTSWe studied 180 consecutive unrelated probands with cardiomyopathy (15 HCM, 15 DCM, and 10 noncompacted) looking for mutations in the TNNT2 gene and in the 4 other main sarcomere genes (MYBPC3, MHY7, TNNI3, and TPM1); 21 probands (11.7%) had pathogenic mutations in TNNT2. Ninety-eight relatives gave consent for clinical examination (mean, 4.7 relatives/family), and 78 gave consent for genetic analysis of TNNT2; a mutation was found in 33 individuals (42%). The total number of probands plus relatives carrying TNNT2 mutations was 54. Assuming that some first-degree relatives who did not undergo genetic study but who did have HCM had the same TNNT2 mutation, the total was 68 patients: 57 affected individuals and 11 asymptomatic carriers.
In 21 families, 6 different mutations were identified: Arg92Gln in 10 families, Arg286His in 5, Arg278Cys in 3, and Arg92Trp, Arg94His, and Ile221Thr in 1 family each. All these variants except 1 (Ile221Thr) had already been published as causes of HCM.8,14,15,17–19
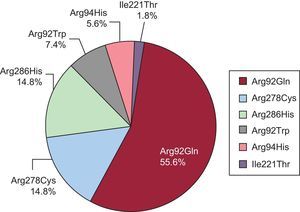
Double mutations were found in 4 probands (19%): 2 with the Arg278Cys mutation (1 with the Arg502Gln mutation in MYBPC3 and 1 with Arg723Cys in MHY7), 1 proband with the Arg286His mutation and the Arg326Gln mutation in MYBPC3, and 1 proband with the Arg92Gln mutation, who was a carrier of a variant in MYBPC3 that could have acted as a genetic modifier. No relatives were carriers of double mutations. The distribution of mutations and patients was as follows (Figure 1): 30 individuals had Arg92Gln (55.6%), 8 had Arg278Cys (14.8%), 8 had Arg286His (14.8%), 4 had Arg92Trp (7.4%), 3 had Arg94His (5.6%), and 1 had Ile221Thr (1.8%).

All affected patients underwent risk stratification for SCD. Thirteen probands (62%), and 100% of families (n = 10) with the Arg92Gln mutation had a history of SCD.
Electrocardiograms and echocardiograms were studied at the first assessment of all mutation carriers, except for 3 patients (because of SCD being the clinical presentation). The electrocardiograms were generally abnormal (voltage criteria for left ventricular hypertrophy with negative T-waves in precordial and inferior leads), sometimes with only mild abnormalities on echocardiography.
At the first assessment, 9 of the patients with the Arg92Gln mutation had a DCM phenotype with severe left ventricular dysfunction. One affected individual with Arg92Gln had a noncompaction cardiomyopathy phenotype. The others had HCM (Tables 1 and 2).
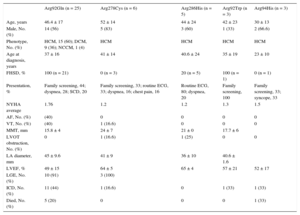
Clinical and Genetic Data on Patients and Families Included in the Study
| Family | TNNT2 mutation | Age/sex | Phenotype | Age at diagnosis, years | FHSD | Presentation | NYHAfunctionnalclass | AF | VT | MMT, mm | LVOT obstruction | LA diameter, mm | LVEF | LGE | PM/ICD (age, years) | Appropriate ICD therapy | Died |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1/Proband | Arg92Gln | 22/M | DCM | 22 | Yes | SCD | I | No | No | 12 | No | No | Yes | ||||
| 1/Mother | Arg92Gln | 70/F | DCM | 60 | Dyspnea | II | Yes | No | 11 | No | 44 | 40 | PM (60) | No | |||
| 1/Sister | Arg92Gln | 42/F | DCM | 42 | SCD | I | Yes | No | 8 | No | No | Yes | |||||
| 2/Proband | Arg92Gln | 31/M | HCM | 13 | Yes | SCD | I | No | Yes | 20 | No | 48 | 60 | ICD (13) | Yes | No | |
| 2/Father | Arg92Gln | 53/M | HCM | 37 | Family screening | II | Yes | Yes | 20 | No | 52 | 55 | ICD (38) | Yes | No | ||
| 2/Aunt | Arg92Gln | 45/F | HCM | 28 | Family screening | III | Yes | No | 22 | No | 50 | 60 | Yes | No | No | ||
| 3/Proband | Arg92Gln | 64/F | DCM | 50 | Yes | Dyspnea | II | Yes | No | 14 | No | 50 | 45 | Yes | No | No | |
| 3/Sister | Arg92Gln | 40/F | HCM | 34 | Family screening | I | No | No | 14 | No | 46 | 60 | Yes | No | No | ||
| 3/Grandson* | Arg92Gln | 8/M | — | — | I | No | No | 7 | No | 25 | 65 | No | No | ||||
| 3/Brother | Arg92Gln | 76/M | DCM | 69 | Dyspnea | IV | Yes | Yes | No | 30 | No | Yes | |||||
| 3/Nephew | Arg92Gln | 45/M | HCM | 35 | Family screening | I | No | No | 17 | No | 33 | 48 | Yes | No | No | ||
| 3/Niece | Arg92Gln | 52/F | HCM | 43 | Family screening | I | No | No | 14 | No | 45 | 60 | No | No | |||
| 3/Grand-nephew | Arg92Gln | 21/M | HCM | 20 | Family screening | I | No | No | 18 | No | 65 | Yes | No | Yes | |||
| 3/Grand-niece | Arg92Gln | 19/F | HCM | 19 | Family screening | I | No | No | 14 | No | 62 | Yes | ICD (19) | No | No | ||
| 4/Proband | Arg92Gln | 72/M | DCM | 67 | Yes | Dyspnea | III | Yes | No | 11 | No | 55 | 30 | Yes | CRT (69) | Yes | |
| 4/Sister | Arg92Gln | 65/F | DCM | 55 | Dyspnea | III | Yes | Yes | 15 | No | 60 | 33 | ICD (63) | ? | No | ||
| 4/Nephew | Arg92Gln | 54/M | DCM | 50 | Dyspnea | III | No | No | No | 30 | CRT (52) | No | |||||
| 4/Grand-nephew | Arg92Gln | 19/M | HCM | 11 | SCD | I | No | Yes | 13 | No | 30 | 65 | ICD (11) | ? | No | ||
| 5/Proband | Arg92Gln | 66/M | DCM | 40 | Yes | Chest pain | II | Yes | Yes | 20 | No | 50 | 30 | CRT + ICD (62) | ? | No | |
| 6/Proband | Arg92Gln | 49/M | HCM | 35 | Yes | Family screening | I | No | Yes | 15 | No | 43 | 60 | Yes | ICD (45) | No | No |
| 6/Sister | Arg92Gln | 54/F | HCM | 24 | Family screening | II | Yes | Yes | 19 | No | 48 | 30 | ICD | ? | No | ||
| 6/Daughter* | Arg92Gln | 7/F | — | — | I | No | No | 7 | No | 27 | 70 | No | No | ||||
| 7/Proband | Arg92Gln | 40/M | HCM | 36 | Yes | Family screening | I | No | No | 22 | No | 47 | 60 | Yes | ICD (38) | No | No |
| 7/Nephew* | Arg92Gln | 12/M | — | — | I | No | No | 8 | No | 28 | 65 | No | No | ||||
| 7/Niece* | Arg92Gln | 6/F | — | — | I | No | No | No | 60 | No | No | ||||||
| 8/Proband | Arg92Gln | 36/M | HCM | 17 | Yes | SCD | I | No | Yes | 20 | No | 33 | 62 | ICD (17) | ? | No | |
| 8/Son* | Arg92Gln | 8/M | — | — | I | No | No | 9 | No | 27 | 70 | No | No | ||||
| 9/Proband | Arg92Gln | 52/F | HCM | 51 | Yes | Dyspnea | II | No | No | 15 | No | 31 | 58 | No | No | No | |
| 9/Daughter | Arg92Gln | 27/F | HCM | 26 | Family screening | I | No | No | 20 | No | 29 | 65 | Yes | No | No | ||
| 10/Proband | Arg92Gln | 46/M | NCCM | 40 | Yes | Routine ECG | III | No | Yes | 10 | No | 60 | 21 | ICD (44) | No | No | |
| 11/Proband | Arg92Trp | 69/F | HCM | 57 | Yes | Family screening | II | No | No | 25 | No | 42 | 33 | ICD (69) | No | No | |
| 11/Son | Arg92Trp | 32/M | HCM | 28 | Family screening | I | No | No | 15 | No | 40 | 65 | No | No | |||
| 11/Daughter | Arg92Trp | 25/F | HCM | 20 | Family screening | I | No | No | 13 | No | 40 | 74 | Yes | No | No | ||
| 11/Daughter* | Arg92Trp | 36/F | — | — | I | No | No | 10 | No | 33 | 65 | No | No | ||||
| 12/Proband | Arg278Cys | 47/M | HCM | 18 | No | Routine ECG | I | No | No | 23 | No | 38 | 60 | Yes | No | No | |
| 12/Father | Arg278Cys | 71/M | HCM | 57 | Routine ECG | I | No | No | 35 | Yes | 58 | 58 | No | No | |||
| 12/Cousin | Arg278Cys | 40/M | HCM | 37 | Family screening | I | No | No | 16 | No | 36 | 62 | No | No | |||
| 13/Proband | Arg278Cys | 41/F | HCM | 37 | No | Dyspnea | I | No | No | 18 | No | 35 | 65 | Yes | No | No | |
| 13/Brother | Arg278Cys | 44/M | HCM | 42 | Family screening | I | No | No | 22 | No | 36 | 70 | Yes | No | No | ||
| 13/Mother* | Arg278Cys | 75/F | — | — | I | No | No | 10 | No | 32 | 60 | No | No | ||||
| 14/Proband | Arg278Cys | 68/M | HCM | 55 | No | Chest pain | II | No | Yes | 30 | No | 44 | 68 | ICD (62) | Yes | No | |
| 14/Daughter* | Arg278Cys | 29/F | — | — | I | No | No | 9 | No | 31 | 62 | No | No | ||||
| 15/Proband | Arg286His | 80/F | HCM | 77 | Yes | Dyspnea | II | No | No | 21 | Yes | 51 | 70 | PM (79) | No | ||
| 15/Son* | Arg286His | 48/M | — | — | I | No | No | 10 | No | 35 | 65 | No | No | ||||
| 16/Proband | Arg286His | 35/M | HCM | 25 | No | Routine ECG | I | No | No | 21 | No | 34 | 60 | No | No | ||
| 17/Proband | Arg286His | 36/F | HCM | 34 | No | Routine ECG | I | No | No | No | No | ||||||
| 18/Proband | Arg286His | 16/M | HCM | 15 | No | Routine ECG | I | No | No | 21 | No | 28 | 65 | No | No | ||
| 18/Mother* | Arg286His | 40/F | — | — | I | No | No | 10 | No | 35 | 60 | No | No | ||||
| 18/Cousin* | Arg286His | 6/M | — | — | I | No | No | No | 75 | No | No | ||||||
| 19/Proband | Arg286His | 53/M | HCM | 52 | No | Routine ECG | I | No | No | 21 | No | 32 | 67 | Yes | No | No | |
| 20/Proband | Arg94His | 15/M | HCM | 14 | Yes | Family screening | I | No | No | 33 | No | 41 | 64 | No | No | No | |
| 20/Father | Arg94His | 39/M | HCM | 34 | Syncope | II | No | No | No | 40 | ICD (34) | ? | No | ||||
| 20/Aunt | Arg94His | 36/F | HCM | 22 | No | Yes | |||||||||||
| 21/Proband | Ile221Thr | 75/F | HCM | 62 | No | Dyspnea | III | Yes | No | 22 | Yes | 50 | 60 | No | No |
AF, atrial fibrillation; CRT, cardiac resynchronization therapy; DCM, dilated cardiomyopathy; ECG, electrocardiogram; F, female; FHSD, family history of sudden death; HCM, hypertrophic cardiomyopathy; ICD, implantable cardioverter-defibrillator; LA, left atrium; LGE, late gadolinium enhancement; LVEF, left ventricular ejection fraction; LVOT, left ventricular outflow tract; M, male; MMT, maximal myocardial thickness; NCCM, noncompaction cardiomyopathy; NYHA, New York Heart Association; PM, pacemaker; SCD, sudden cardiac death; VT, sustained or nonsustained ventricular tachycardia.
Clinical, Echocardiographic, and Prognostic Characteristics of Patients with Cardiomyopathy According to the TNNT2 Mutation
| Arg92Gln (n = 25) | Arg278Cys (n = 6) | Arg286His (n = 5) | Arg92Trp (n = 3) | Arg94His (n = 3) | |
|---|---|---|---|---|---|
| Age, years | 46.4 ± 17 | 52 ± 14 | 44 ± 24 | 42 ± 23 | 30 ± 13 |
| Male, No. (%) | 14 (56) | 5 (83) | 3 (60) | 1 (33) | 2 (66.6) |
| Phenotype, No. (%) | HCM, 15 (60); DCM, 9 (36); NCCM, 1 (4) | HCM | HCM | HCM | HCM |
| Age at diagnosis, years | 37 ± 16 | 41 ± 14 | 40.6 ± 24 | 35 ± 19 | 23 ± 10 |
| FHSD, % | 100 (n = 21) | 0 (n = 3) | 20 (n = 5) | 100 (n = 1) | 0 (n = 1) |
| Presentation, % | Family screening, 44; dyspnea, 28; SCD, 20 | Family screening, 33; routine ECG, 33; dyspnea, 16; chest pain, 16 | Routine ECG, 80; dyspnea, 20 | Family screening, 100 | Family screening, 33; syncope, 33 |
| NYHA average | 1.76 | 1.2 | 1.2 | 1.3 | 1.5 |
| AF, No. (%) | (40) | 0 | 0 | 0 | 0 |
| VT, No. (%) | (40) | 1 (16.6) | 0 | 0 | 0 |
| MMT, mm | 15.8 ± 4 | 24 ± 7 | 21 ± 0 | 17.7 ± 6 | |
| LVOT obstruction, No. (%) | 0 | 1 (16.6) | 1 (25) | 0 | 0 |
| LA diameter, mm | 45 ± 9.6 | 41 ± 9 | 36 ± 10 | 40.6 ± 1.6 | |
| LVEF, % | 49 ± 15 | 64 ± 5 | 65 ± 4 | 57 ± 21 | 52 ± 17 |
| LGE, No. (%) | 10 (91) | 3 (100) | |||
| ICD, No. (%) | 11 (44) | 1 (16.6) | 0 | 1 (33) | 1 (33) |
| Died, No. (%) | 5 (20) | 0 | 0 | 0 | 1 (33) |
AF, atrial fibrillation; DCM, dilated cardiomyopathy; ECG, electrocardiogram; FHSD, family history of sudden death; HCM, hypertrophic cardiomyopathy; ICD, implantable cardioverter-defibrillator; LA, left atrium; LGE, late gadolinium enhancement; LVEF, left ventricular ejection fraction; LVOT, left ventricular outflow tract; MMT, maximal myocardial thickness; NCCM, noncompaction cardiomyopathy; NYHA, New York Heart Association; SCD, sudden cardiac death; VT, sustained or nonsustained ventricular tachycardia.
In the analysis of the 57 individuals with cardiomyopathy, the mean age of presentation was 37 ± 17 years; 30 (56%) were male. The initial symptoms were dyspnea in 10 individuals (24%), SCD in 5 (12%), chest pain in 2 (5%), and syncope in 1 (2%); the others were asymptomatic and diagnosis was made following familial screening in 17 (40%) and routine electrocardiography in 7 (17%). Complications during follow-up consisted of ventricular tachycardia or ventricular fibrillation in 3 individuals (7%), heart failure in 17 (40%), syncope in 6 (14%), chest pain in 5 (12%), stroke in 3 (7%), and atrial fibrillation in 11 (24%).
The mean MMT was 18.4 ± 6 (8-35) mm. Patients with the Arg92Gln mutation had a mean MMT of 15.8 ± 4mm. Only 3 (7%) had left ventricular outflow tract obstruction > 30mmHg (1 patient each with the Arg278Cys, Arg286His and Ile221Thr mutations). There was a high prevalence of left ventricular systolic dysfunction, which was present in 12 patients (31%): 10 with the Arg92Gln mutation, 1 with Arg92Trp, and 1 with Arg94His. The mean left atrial diameter was 43 ± 9mm. Cardiac magnetic resonance was performed in 17 patients, 15 of whom (88%) had extensive late gadolinium enhancement.
All patients were followed up (mean, 5 ± 2.5 years). No significant changes were observed in cardiac dimensions or systolic function, independently of cardiac phenotype (HCM, DCM, or noncompaction).
Fourteen individuals (33%) had an ICD implanted: 8 probands (6 as primary prevention and 2 as secondary prevention) and 6 relatives (5 as primary prevention and 1 as secondary prevention). Of 14 patients with ICD, 11 had the Arg92Gln mutation; 1 had Arg92Trp; 1 had Arg278Cys (double mutation in MYBPC3), and 1 had Arg94His.
Three patients with Arg92Gln required a biventricular device for congestive heart failure, and 2 required a dual chamber pacemaker due to sinus dysfunction or atrioventricular block. No patients required myectomy or cardiac transplantation.
During follow-up, 3 patients died: 1 due to heart failure at 60 years (patient with severe left ventricular dysfunction and dilatation, Arg92Gln), another due to stroke (Arg94His), and the third of unknown cause (patient with chronic heart failure and ventricular dysfunction, Arg92Gln phenotype and relatives with Arg92Gln mutation, but no genetic confirmation).
Three patients had at least 1 appropriate ICD shock: 2 with Arg92Gln and 1 with Arg278Cys (this patient had a double mutation in MYBPC3).
Regarding the 11 deaths related to TNNT2 mutations—the total of SCDs from the pedigree analysis (n = 6), SCDs as first presentation of the disease (n = 3), and the deaths during follow-up (n = 2)—6 (54.5%) were related to sport, 9 (81.8%) were male, and the mean age was 21.7 ± 10.9 years. Seven (63.6%) had an HCM phenotype, 2 (18.2%) had a DCM phenotype, and 2 (18.2%) had an unknown phenotype. The mean MMT was 14.6 ± 5mm. Two of the patients who died had had an episode of atrial fibrillation. In the Arg92Gln group, 100% of the group had a family history of SCD vs 28.6% of the group with other TNNT2 mutations distinct from Arg92Gln (P = .008). Sudden cardiac death was the first presentation of disease in 6 patients from the Arg92Gln group; no patients in the group without Arg92Gln had SCD as the first presentation of disease. Also, 3 recoveries from SCD were documented in the group with Arg92Gln and none were documented in the group without. Regarding appropriate ICD therapies, these occurred in 3 patients from the group with Arg92Gln and 1 in the group without. During follow up, 2 patients from the Arg92Gln group died; there were no deaths related to other mutations.
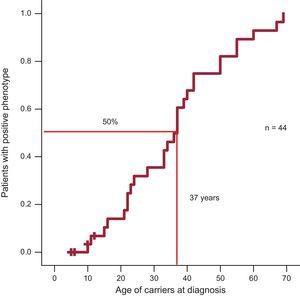
Regarding penetrance, 50% of individuals in the Arg92Gln group had a positive phenotype at 37 years (Figure 2).

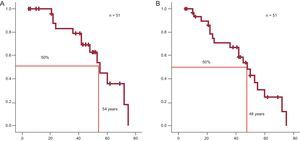
Survival from birth was calculated for carriers of TNNT2 mutations, including relatives identified from a family tree who had HCM or were obligate carriers. The Arg92Gln mutation was associated with a higher rate of SCD at a young age. The mean survival of patients with Arg92Gln was 54 years (95% confidence interval, 46-62 years), but decreased to 48 years if patients with recovery from SCD or appropriate ICD shock were included (Figure 3). In the group with Arg92Gln, survival at 55 years was only 50% (95% confidence interval, 41%-58%).
. B: As in A, but including recovery from sudden cardiac death and patients with appropriate therapies from implantable cardioverter defibrillator.")
A: Sudden cardiac death-free survival of all individual carriers of the Arg92Gln mutation (including relatives with hypertrophic cardiomyopathy and obligate carriers). B: As in A, but including recovery from sudden cardiac death and patients with appropriate therapies from implantable cardioverter defibrillator.
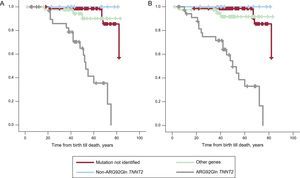
The low rate of SCD hampered subgroup analysis. However, when probands were compared with relatives, there was no difference in cardiac mortality (Kaplan-Meier log rank test, P = .62). Survival curves of the study population were compared: the Arg92Gln mutation was compared against the 5 other TNNT2 mutations (due to the different prognostic profile previously mentioned), against other sarcomeric genetic mutations, and against patients with no identified mutation (Figure 4). Analysis of the survival curve showed that patients with the Arg92Gln mutation had a much lower survival rate than other groups (other TNNT2 mutations, other sarcomeric gene mutations, and patients with no identified mutation). There were statistically significant differences from the group with other TNNT2 mutations (log rank test, 11.71; P = .0006). Also, the group of patients with TNNT2 mutation generally had a worse survival rate than the group with other gene mutations or no identified mutations, but this was due to the patients with the Arg92Gln mutation. Patients with double mutations were not analyzed together with other individual mutations.

A: Sudden cardiac death-free survival in the 4 groups of patients with hypertrophic cardiomyopathy according to genetic result. B: Sudden cardiac death-free survival including recovery from sudden cardiac death and patients with appropriate implantable cardioverter-defibrillator therapies.
Mutations in TNNT2 are considered an infrequent cause of HCM (5%).8,9,20,21 In this study, the prevalence of TNNT2 mutations was higher than in previous series,9,10,17,18,22–24 partly because of a probable founder effect. This study had the highest number of families, and one of the broadest longitudinal cohorts of TNNT mutations (Table 3) since the first published study to show a high incidence of SCD in such patients.9 Our results confirm that SCD is common in young people with some mutations of this gene, but this finding cannot be extrapolated to all known mutations.
Published Studies on Survival With Mutations in the TNNT2 Gene
| Study | No. of families | No. of patients | No. of cardiac deaths | No. of sudden deaths | Mutations |
|---|---|---|---|---|---|
| Watkins et al9 | 11 | 112 | 50 | 39 | Ile79AsnArg92GlnPhe110IleΔGlu160Glu163LysGlu244AspIntron 15 G>A Arg278Cys |
| Nakajima-Tanaguchi et al22 | 1 | 4 | 2 | 2 | Ala104Val |
| Moolman et al10 | 2 | 22 | 7 | 7 | Arg92Trp |
| Anan et al17 | 6 | 18 | 2 | 2 | Phe110Ile |
| Torricelli et al18 | 5 | 10 | 0 | 0 | Phe110IleArg130CysΔGlu160Arg92GlnArg278Cys |
| Pasquale et al23 | 20 | 92 | ? | 7 | Arg278CysArg92LeuArg92TrpΔGlu163IVS15+1G>AAla104Val Arg278HisArg92Gln Arg94LeuGlu163LysGlu83LysIle79Asn |
| Ripoll-Vera et al, 2015* | 21 | 54 | 11 | 6 | Arg92GlnArg92TrpArg286HisArg278CysArg94HisIle221Thr |
Electrocardiography was almost always abnormal, sometimes with only mild abnormalities on echocardiography, which underscores the role of both investigations in detecting this disease, something which has previously been expressed, but is important to reiterate.
In comparison with previous studies,9,10,17,18,22–24 there was a lower prevalence of left ventricular outflow tract obstruction at rest, which was present in only 7% of patients, somewhat similar to the most recent published study.23
We identified 6 studies on mutations in TNNT2 that analyzed survival.9,10,17,18,22,23 They comprised a total of 258 carriers and 68 deaths apparently of cardiovascular causes, mostly sudden, but 50 of those deaths were from only 1 publication. Therefore, most of those studies had a low number of SCDs, which coincides with the prospective data from this study (Table 3).
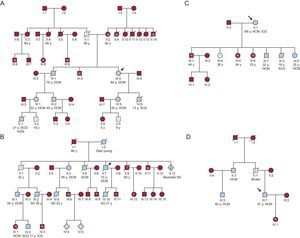
Family tree analysis (Figure 5) showed a high prevalence of SCD in affected families, but only with some mutations (mainly Arg92Gln, but also Arg92Trp and Arg94His). The SCD rate was too small to perform adequate statistical analysis of subgroups, as was the case in previous studies.23 The different prognoses in families with the same mutations indicates that other mechanisms (genetics, epigenetics, or environmental factors) could have had an influence.
, 4-Arg92Gln- (B), 11-Arg92Trp- (C) and 12-Arg278Cys- (D) families. DCM, dilated cardiomyopathy; HCM, hypertrophic cardiomyopathy; ICD, implantable cardioverter-defibrillator; SCD, sudden cardiac death; SD, sudden death; y, years.")
Previous studies24 have already documented cases of SCD with little or even no hypertrophy in carriers of TNNT2 mutations. This was also observed in some of our patients, but only with the Arg92Gln mutation. In patients who had SCD, HCM was more common than DCM, and MMT was only 14.6 ± 6mm. Because of the relative lack of events in the present cohort, the relative risk of SCD could not be determined in mutation carriers who had normal echocardiograms.
This study has detected important prognostic differences between patients with 1 of the TNNT2 mutations (Arg92Gln) and those with the 5 other TNNT2 mutations, other sarcomeric gene mutations, and no identified mutation. Compared with other groups, patients with the Arg92Gln mutation had an earlier presentation and worse prognosis. They had a high incidence of SCD, a mixed phenotype (HCM with mild hypertrophy, DCM with ventricular dysfunction, noncompaction cardiomyopathy), absence of obstruction, significant fibrosis, and frequent need for ICD or resynchronization therapy.
Progressive thinning of the myocardium and deterioration in contractile function are a well-known phenomenon of HCM.3,4 In this study, 9 patients already had ventricular dilatation at presentation (age, 50.6 ± 14.6 years, all with Arg92Gln) and 2 died of heart failure during follow-up, indicating that progression of heart failure could be relatively common in such patients. In fact, the patients with HCM were younger (28.8 ± 11.8 years) than those with DCM. However, there were no cases of progressive ventricular dilatation. This could have been due to the short follow-up, and therefore we cannot conclude with complete certainty that patients with DCM at diagnosis really were patients with HCM in burn-out phase.
One proband with Arg92Gln had noncompaction cardiomyopathy, an association that had not been described until now. It is important to remember that the TNNT2 gene should always be included in genetic study in patients with HCM and DCM, and possibly also, based on our findings, in cases of noncompaction cardiomyopathy.
The Arg92Trp mutation affects the same amino acid as Arg92Gln, and therefore it behaves similarly. It is associated with HCM, mild hypertrophy, and a high incidence of SCD.5,19
The Arg278Cys mutation is also associated with late-presentation HCM and mild-to-moderate hypertophy.9,18,25–27 Sudden cardiac death is uncommon in young people but is common in patients of advanced age. It is often associated with other pathogenic variants.27 From the 3 families with the Arg278Cys mutation, 2 probands had double mutations. There was no history of SCD, and the MMT was highly variable (14-35mm).
The Arg286His mutation has been associated with HCM.19 The 3 families with this mutation had HCM with an MMT of 21mm, without a high risk of SCD.
The Arg94His mutation is also a known cause of HCM. The first symptoms can manifest in childhood and patients may also have severe arrhythmic events with an apparently mild phenotype.28
Finally, the Ile221Thr mutation had not previously been published. It was considered a rare genetic variant affecting 1 relevant functional region. It could only be studied in the index case (with clear HCM), and therefore it was not possible to study its segregation in the family to confirm its pathogenicity.
Double mutations could confer a worse prognosis. Other studies on mutations in the TNNT2 gene did not offer data on additional mutations on other genes. In this series, double mutations were found in 4 probands (19%), but no double mutations were found in the relatives studied. In the literature, double mutations are found in only 5% of cases of HCM.7,23 Only 1 of our 4 patients with a double mutation had an unfavorable risk stratification, and had ICD implantation, but there were insufficient data to establish a prognosis relating to double mutations.
A probable founder effect was demonstrated for the Arg92Gln mutation. Of the 10 families, 9 originated from the same village. Using extensive genealogical study (parish archives and population census), we found that 6 of these families had a common ancestor born in 1784. Haplotype study was not performed as it was not available in our setting.
LimitationsAs with previous studies, there could have been selection bias because the study was carried out in a specialized unit. Patients with genetic and clinical (or pathological) confirmation were included, as were relatives with a positive phenotype and those who had sudden death, who were also presumed to be affected despite not having genetic confirmation.
The phenotypic differences found between some patients with the same mutations shows that the prognosis of these individuals is influenced by many factors other than the mutation itself.
CONCLUSIONSInvestigation of the genotype-phenotype correlation in HCM remains a challenge. Mutations in the TNNT2 gene were more common in our series than in previous studies, partly because of a probable founder effect. The clinical and prognostic profiles depended greatly on the mutation. Risk profile was significantly worse in carriers of Arg92Gln than in other patients. Sudden cardiac death was a frequent complication and can occur in young individuals with little or no hypertrophy. Dilated cardiomyopathy with ventricular dysfunction was fairly common among carriers of some mutations (Arg92Gln).
Overall, these findings have important implications for the clinical and genetic study of families with cardiomyopathy, above all the finding of the Arg92Gln mutation, which, given its demonstrated malignancy, should cause a change in the management of individuals in SCD prevention.
FUNDINGRed de Investigación Cardiovascular del Instituto de Salud Carlos III (RD12/004/0069) and CIBEROBN (Centro de Investigación Biomédica en Red de la Fisiopatología de la Obesidad y la Nutrición) (CB12/03/30038), Madrid, Spain.
CONFLICTS OF INTERESTNone declared.