Keywords
INTRODUCTION
Pulmonary hypertension (PH) is defined1 as an increase in mean pulmonary arterial pressure(PAP) >25 mmHg at rest as determined by rightheart catheterization (RHC). Currently, the normal behavior of pulmonary pressure on exercise remainsunknown, and it presents wide variability accordingto age and the degree of physical fitness in the healthyindividual. Thus, a definition of PH on exercisecannot be established.
CLINICAL CLASSIFICATION OF PULMONARYHYPERTENSION
Pulmonary hypertension can be found in differentclinical conditions3,4 and are classified into 5 groups:group 1, pulmonary arterial hypertension (PAH);group 2, pulmonary hypertension associated withleft heart disease (PHLHD); group 3, pulmonaryhypertension associated with lung disease orhypoxemia; group 4, chronic thromboembolic pulmonary hypertension (CTEPH); and group5, pulmonary hypertension with unclear ormultifactorial mechanisms.
This classification1 (Table 1) is based on clinicaldata, and groups together the different processes anddiseases that share pathophysiological mechanisms,clinical presentation and therapeutic approaches.In relation to previous classifications, substantialmodifications have been made to group 1. The termfamilial PAH has been replaced by heritable PAH,since specific gene mutations have been identified insporadic cases with no family history of the disease.Among the heritable forms of PAH are sporadicidiopathic PAH (IPAH) with germline mutationsand clinical cases with a family background with orwithout identified mutations. This new category ofheritable PAH does not require genetic testing, asthis would not change its clinical management.

The classification of congenital heart diseaseunderlying PAH has been updated to include a clinicalversion (Eisenmenger's syndrome, PH associatedwith systemic-to-pulmonary shunt, PH associatedwith small defects and PH after shunt repair) and ananatomical-pathophysiological version (Table 2) tobetter define each patient.

It remains difficult to classify pulmonaryveno-occlusive disease and pulmonary capillaryhemangiomatosis in group 1, because they sharesome characteristics with IPAH, but also presentsome differences. Finally, it was decided to includethem in a different, but not entirely separate, categoryto that of PAH, and thus have been denominated asclinical group 1'.
PATHOBIOLOGY OF PULMONARY ARTERIALHYPERTENSION
Pulmonary arterial hypertension is clinicallydefined as a group of diseases characterized by agradual increase in pulmonary vascular resistanceleading to right ventricular failure and early death.5 Prognosis is related to complex pathophysiological interactions between the progression (or regression)rate of the obstructive changes in pulmonarymicrocirculation and the response of the overloadedright ventricle (RV). The known main prognosticfactors in this disease are derived from rightventricular function (hemodynamic, clinical andbiochemical). The increase in afterload remainsthe main determinant of heart failure in patientswith PAH and CTEPH, because its elimination asan outcome of lung transplantation or pulmonary endarterectomy almost invariably leads to rapidrecovery of RV function.
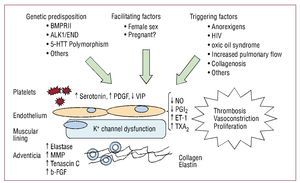
The pathophysiological basis of the increase inpulmonary vascular resistance is hypertensive vasculardisease in small arteries and pulmonary arterioles.Multiple cellular and molecular factors are involvedin its development that lead to remodelling of thevessel wall via 4 basic mechanisms: vasoconstriction,cell proliferation, thrombosis, and immune factors.Its origin is unknown, but a genetic predispositionfacilitated and triggered by factors that lead to diseaseonset6 has been suggested (Figure 1).

Figure 1. Pathobiological mechanisms in the development of pulmonary arterial hypertension. ALK1 indicates activin-receptor-like kinase-1; BMPR2, bone morphogenetic protein receptor 2; ENG, endoglin; ET-1, endothelin 1; b-FGF, basic fibroblast-derived growth factor; 5-HTT, serotonin transporter gene; MMP, metalloproteinases; NO, nitric oxide; PDGF, platelet-derived growth factor; PGI2, prostacyclin; TXA2, thromboxane A2; HIV, human immunodeficiency virus; VIP, vasoactive intestinal peptide.
The different molecular mediators involved in thedevelopment of the disease, their main mechanismof action, and cell type involved are summarizedin Table 3. The final outcome is a predominanceof the mediators that lead to vasoconstriction, cellproliferation, and vascular thrombosis versus thosewhich perform the opposite function. Knowledgeof these mediators is not only important tounderstanding the natural history of the disease, butalso because they are the target of different currenttreatments as well as being a research focus.

Genetics of Pulmonary Arterial Hypertension
Recently,7 significant progress has been madein this field, especially in the study of BMPR2(bone morphogenetic protein receptor 2), ALK-1 (activin receptor-like kinase 1) and 5-HTT(endoglin associated with hereditary hemorrhagictelangiectasia and the serotonin transporter gene),whose LL polymorphism (2 long alleles) seemsto be more common in patients with PAH than incontrols.
The BMPR2 Gene
This gene codes for a membrane receptor belongingto the transforming growth factor b (TGF-b) receptorfamily. It is expressed in pulmonary endothelium,smooth muscle cells, and macrophages, and regulatesmultiple cell functions: proliferation, migration,differentiation, and apoptosis. Its mutation givesrise to haploinsufficiency, that is, low amountsof the receptor, which leads to increased cellularproliferation and the inhibition of cell apoptosis.The gene is located on the long arm of chromosome2 (2q31,32), it has 13 exons, and up to 298 differentpoint mutations have been described.2
In familial PAH, currently classified as heritable,mutations have been described in up to in 70%of cases, with autosomal dominant inheritance.Penetrance is incomplete and only 20% of thesecarriers will contract the disease. Another featureis the phenomenon of genetic anticipation, that is,subsequent generations will develop the disease atyounger ages. Mutations have been described inapproximately 20% of cases with idiopathic PAH,in 18% with anorexigen-associated PAH, and in 6%in PAH associated with congenital heart disease.In small series of cases of PAH associated withcollagenosis, human immunodeficiency virus (HIV),and toxic oil syndrome, no mutations were found onthe BMPR2 gene.7-9
Patients carrying the mutated gene differ insome respects from those who do not carry it:the disease appears at a younger age, they have apoorer hemodynamic profile and are less likely to respond to an acute vasodilator test; however, thereare no differences in survival rates and clinicalmanifestations at diagnosis.10
EPIDEMIOLOGY OF PULMONARYHYPERTENSION
Currently, there are no comparativeepidemiological data on the prevalence of thedifferent groups of PH. In an echocardiographicstudy,11 the prevalence of PH (defined by systolic PAP>40 mmHg) in 4579 patients was 10.5%. Of the483 patients with PH, 78.7% had left heart disease(group 2), 9.7% had lung disease and hypoxemia(group 3), 4.2% had PAH (group 1), and 0.6%had CTEPH (group 4); it was not possible reach adiagnosis in the remaining 6.8%.
Group 1
Four national registries12-15 were recently conducted and described the epidemiology of PAH. The lowest estimated prevalence of PAH and IPAHis 15 cases/million and 5.6 cases/million, respectively,and the highest is 26 cases/million and 9 cases/million, respectively. The lowest estimated incidenceof PAH was 2.4 cases/million/y and the highestwas 7.6 cases/million/y. The proportion of womento men was around 2:1, and mean age at diagnosiswas approximately 50 years, with an increasingnumber of patients older than 70 years (10%-17%according to the registries). In the Spanish Registryof Pulmonary Hypertension (Registro Español deHipertensión Pulmonar; REHAP),15 34% of thepatients had IPAH and 3% had a family historyof PAH. In the associated PAH subgroup, 16%had connective tissue disease (especially systemicsclerosis), 17.5% had congenital heart disease, 6.4% had portal hypertension, and 5.9% had HIVinfection.
It is believed that PAH is much more prevalentin emerging countries,16 where relatively commondiseases such as schistosomiasis, sickle-cell anemia,HIV infection and congenital heart disease may becomplicated by PAH.
Group 2
Pulmonary hypertension associated with leftheart disease is the most frequent cause of PH. Heartfailure is a serious and common disease in westerncountries and its incidence in people older than65 years is approximately 10/1000 population/y.17 In 44% of the patients, left ventricular ejectionfraction (LVEF) is normal, heart failure is causedby diastolic dysfunction, and is accompanied byPH in up to 83% of the patients, according to arecently published population study.18 In 45% of the patients, heart failure is caused by systolicdysfunction and is accompanied by PH in 60% ofthe patients.
Group 3
Pulmonary hypertension due to lung diseaseor hypoxemia. In advanced chronic obstructivepulmonary disease, PH is highly prevalent (>50%),although in general it is only of moderate severity.19 The prevalence of PH is 32%-39% in interstitiallung disease. Combined pulmonary fibrosis andemphysema is associated with a higher prevalenceof PH.20
Group 4
The incidence of CTEPH after pulmonaryembolism remains uncertain, although most expertsbelieve that it is 0.5%-2%.1,19 In approximately 40%-50% of the patients with CTEPH there is no clinical event compatible with deep venous thrombosis orpulmonary embolism.
The only population registry that includes CTEPHis REHAP.15 This registry reports an incidence of 0.9cases/million/y and a prevalence 3.2 cases/million.Of the patients with PH included in this registry,15% had CTEPH.
DIAGNOSIS OF PULMONARYHYPERTENSION
Diagnosis is a step-wise process that begins withsuspected PH, followed by confirmation of thediagnosis,21 identification of the specific etiology(IPAH should be considered a diagnosis by exclusion)and finally assessment of severity (using clinical,echocardiographic, and hemodynamic parameters,biomarkers and exercise capacity), a key aspect inthe choice of treatment and follow-up.
The diagnostic algorithm (Figure 2) begins byidentifying the more common clinical groups of PH1 (group 2, left heart disease; group 3, lung disease),then distinguishes group 4 (CTEPH) and finallymakes the diagnosis and recognizes the different typesin group 1 (PAH) and group 5 (miscellaneous).

Figure 2. Diagnostic algorithm of pulmonary hypertension. ANA indicates antinuclear antibodies; RHC, right heart catheterization; TTE, transthoracic echocardiography; TEE, transesophageal echocardiogram; PVOD, pulmonary veno-occlusive disease; PAH, pulmonary arterial hypertension; PCH, pulmonary capillary hemangiomatosis; PH, pulmonary hypertension; PAPm, mean pulmonary arterial pressure; PWP, pulmonary wedge pressure; PFT, pulmonary function test; MRI, magnetic resonance imaging; HRCT, high-resolution computed tomography; V/Q, ventilation/perfusion; HIV, human immunodeficiency virus. Adapted from Galiè N, et al.1 x by permission of the author, publishers and the European Society of Cardiology.
Clinical suspicion of PH is based on symptoms,the presence of risk factors, the findings ofphysical examination and the results of simpleexaminations such as chest radiograph and ECG. Ifthe initial assessment confirms the suspicion of PH,transthoracic echocardiogram, pulmonary functiontests and high-resolution computed tomography(CT) of the chest are performed to identify lungdisease (group 3) or left heart disease (group 2). Ifthere is no evidence of heart or respiratory diseaseor PH seems "disproportional" to the severity of theunderlying disease, a ventilation/perfusion lung scan(V/Q) is recommended. If the V/Q lung scan showsmultiple segmental perfusion defects, CTEPHshould be suspected. The final diagnosis of CTEPHrequires CT pulmonary angiography, RHC, andselective pulmonary angiography. If this possibilityis ruled out, and once PH is confirmed by RHC, thedifferent types of PAH are investigated.
Some aspects relevant to diagnosis are describedin the following sections.
Clinical Presentation
The initial symptom is progressive effortdyspnea. When RV dysfunction progresses,angina or effort syncope may appear due to theinability of the RV to adapt cardiac output toexercise; these symptoms occur at rest only inadvanced phases. In populations at risk1-4 of PAH(congenital heart disease, history of pulmonaryembolism, connective tissue disease [CTD], HIV and exposure to toxic substances associated withPH), the presentation of these symptoms requiresconfirmation by echocardiogram. In the absenceof symptoms an echocardiogram be conductedonly in patients with scleroderma, liver transplantcandidates, and in the relatives of patients withheritable PAH.
Transthoracic Echocardiography
This should be performed whenever PH issuspected. It provides an estimate of pulmonaryartery systolic pressure (PSP), left ventricularsystolic and diastolic function and valve disease anddetects systemic-to-pulmonary shunt (with the useof agitated saline).
The calculation of PSP is based on the simplifiedBernoulli equation, in which PSP = 4 x (maximumtricuspid regurgitation velocity)2 + right atrial pressure(RAP). The RAP can be calculated using the diameterand the respiratory variation of the inferior vena cava,although a fixed value of 5 mmHg or 10 mmHg is oftenassumed. When it is difficult to measure peak tricuspidregurgitation velocity, the use of intravenous agitatedsaline is recommended as this significantly improvesthe Doppler ultrasound signal.
In general, the correlation between PSP estimatedby echocardiogram and by RHC is good (0.57-0.85). However, when PSP is determined byechocardiography the hemodynamic value can beoverestimated by >10 mmHg in up to 48% of patients,especially if the Doppler ultrasound recording is ofpoor quality.22 Furthermore, tricuspid regurgitationis found in approximately 80% of patients withPSP >35 mmHg and the ability to analyze flowvaries according to the underlying disease. Thus, ina study of 374 patients with pulmonary disease, arecording of sufficient quality to estimate PSP wasonly obtained in 44%.23
The PSP values vary according to the age andweight of the patient.24 Thus, PSP >40 mmHg isfound in 6% of individuals older than 50 years andin 5% of those with a body mass index of 30.
In view of this, PH cannot be accuratelycharacterized using a PSP cut-off point based onDoppler ultrasound.
Other echocardiographic variables shouldalways be considered in patients with suspectedPH1: right heart chamber dilatation (inferiorvena cava, right atrium [RA], RV and pulmonary artery), the flattening or inversionof the interventricular septum toward the LV,midsystolic collapse and a pulmonary flowacceleration time <80 ms increase the likelihoodof the patient having significant PH.
Table 4 and Table 5 show the probability of adiagnosis of PAH based on echocardiographiccriteria, symptoms, and risk factors, according tothe current European clinical practice guidelines.1


If systemic-to-pulmonary shunt is detectedduring diagnostic study with agitated saline orPAH is suspected on the basis of clinical findings,a transesophageal echocardiogram is recommendedto confirm the diagnosis. Complementary imagingtechniques, such as magnetic resonance imaging,may be required in some cases especially if thesystemic-to-pulmonary shunt is extracardiac or incomplex congenital heart disease.25
If diastolic dysfunction is suspected as the originof PH with preserved LV systolic function in theechocardiographic study, a complete study ofdiastolic function26 of mitral valve flow, mitralannular flow and pulmonary vein flow by pulsed andtissue Doppler imaging is recommended. Similarly,the presence of left atrial dilatation and the degree ofLV hypertrophy should be assessed.
Ventilation/Perfusion Lung Scan
This is the method of choice to exclude CTEPHduring the systematic study of patients with PH. Anormal- or low-probability V/Q scan1,25 effectively excludes CTEPH with a sensitivity between 90% and100% and a specificity between 94% and 100%.
High-Resolution Computed Tomography
This is recommended at the time of initial diagnosisof patients with PH.1 Computed tomographyprovides detailed images of lung parenchyma andfacilitates accurate diagnosis of the interstitial lungdisease (ILD) and emphysema. In patients with PAHassociated with CTD and who present significantevidence of ILD, CT assesses the extent of thevascular disease and the possible fibrosis associatedwith the immune disease.
Computed tomography is essential whenthere is clinical suspicion of pulmonary venoocclusive disease (PVOD) or pulmonary capillaryhemangiomatosis.27 Centrilobular ground-glassopacities (diffuse centrilobular nodular opacity),subplural thickened septal lines and mediastinallymphadenopathies are characteristic of PVOD.
Spiral CT Angiography (CT PulmonaryAngiogram)
This should be conducted in patients in whomV/Q scan is suggestive of CTEPH.
Right Heart Catheterization
Right heart catheterization is required todiagnose PAH, assess the severity of hemodynamic deterioration, and analyze the vasoreactivityof the pulmonary circulation.1,4,28 Right heart catheterization procedures have low rates ofmorbidity (1.1%) and mortality (0.055%) whenconducted in specialized centers.
The following variables should be recorded: PAPtest (systolic, diastolic and average), right atrialpressure, pulmonary wedge pressure (PWP), andRV pressure. If possible, cardiac output should bemeasured in triplicate by thermodilution or by theFick method (this is obligatory when systemic-to-pulmonary shunt is present). Similarly, superiorvena cava, pulmonary artery, and systemic arterialblood oxygen saturations and pulmonary vascularresistance should be measured.
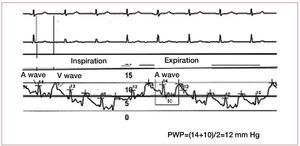
Care is needed when determining PWP, sincea value <15 mmHg is required to establish thediagnosis of PAH. However, the PWP measurementmethod is not standardized and this has led towide inter-observer variability.29 Some groupsconduct measurements at the end of expiration,others take measurements during an apneic pause,and yet others rely on automatic measurementsprovided by software programs. Intrathoracicpressures have almost no effect on intracardiacpressures at the end of expiration, and this is thecorrect moment to measure PWP. In addition, thecatheter should be correctly positioned to guarantee good transmission of left atrial pressure throughthe pulmonary capillary bed. The A and V wavesshould be examined to ensure that they are clearlydifferentiated, and oximetry performed at the distalend of the catheter to confirm correct positioning(the O2 saturation data obtained is similar to thatof arterial O2 saturation). Occasionally, the cathetershould be advanced to a more distal position in thepulmonary vessel to obtain a better tracing.
The A wave is produced at the time of atrialcontraction. Retrograde transmission of the wavecan be observed on the PWP tracing and appearsdelayed at the end of QRS or immediately after this(Figure 3) in the ECG. The maximum and minimumA wave values are measured just before inspiration,and the PWP is the average of these values. TheV wave is produced when blood fills the atriumand the mitral valve is closed. This is observed inthe PWP recording after the T wave in the ECG.A very prominent V wave can hinder the accuratemeasurement of PWP and tends to appear due tosevere mitral regurgitation or severe alterations inLV distensibility.

Figure 3. Pulmonary wedge pressure (PWP) tracing showing respiratory variations and the temporal relationship of the A and V waves on ECG.
Given the steady increase in the age of patientswith PH and the great number of comorbiditiesconsidered as typical risk factors of diastolic LVdysfunction, the reliable differential diagnosisof PAH and PHLHD has become increasinglyimportant and complex, despite good techniquesfor measuring PEP.19,21 Thus, the Registry to Evaluate Early And Long-term PAH DiseaseManagement (REVEAL),14 is being conducted in the USA. It included 2525 patients (mean age, 53 [14] years) with PAH (PWP<15 mmHg). Of these,40% had systemic hypertension; 33%, obesity; 21%,sleep apnea; 12%, diabetes mellitus; and 4.5%,kidney failure. Patients were older than in previousregistries and had a high number of risk factors fordiastolic heart failure.
A hemodynamic study was recently conducted in3920 patients with PH,30 and analyzed the reliabilityof PWP to distinguish PAH and PHLHD comparedto LV end-diastolic pressure (the gold standard ofLV preload). Approximately half of the patientsclassified as having PAH based on PWP <15 mmHg,actually have PHLHD when based on the criterionof an LV end-diastolic pressure <15 mmHg. Thus, ifthe patient presents a clinical profile compatible withPHLHD19 (Table 6), a direct measurement of LVend-diastolic pressure is recommended to confirmthe diagnosis of PAH if PWP is <15 mmHg.

In some patients with high clinical suspicion ofPHLHD who have received diuretics, low PWP andLV end-diastolic pressure values may be observed.To confirm the diagnosis of PAH, RHC withvolume or exercise challenge is recommended.1,5 These procedures have not been standardized andeach cardiac catheterization laboratory has its ownprotocol. Catheterization with volume challenge issimpler and basically consists in the perfusion of1000 ml of physiological saline solution over 20 min,taking measurements every 250 ml. Challenge isinterrupted when PWP is >18 mmHg or symptomsappear.
Pulmonary Vasoreactivity Testing
This should be conducted at the time of diagnosisto identify patients who may benefit from treatmentwith calcium channel blockers. The test shouldbe conducted with short-acting, safe and easy toadminister drugs that have few or no systemiceffects. Currently, the most commonly used agentis nitric oxide; although there is wide experiencewith intravenous epoprostenol and intravenousadenosine, these have an increased risk of causingsystemic vasodilator effects.
A positive acute response1,4,28 (positive acute responder) is defined as a reduction in mean PAP>10mmHg to reach an absolute value of mean PAP<40mmHg with unchanged or increased cardiac output.Only 10% of patients with IPAH will fulfill thesecriteria. Only 50% of IPAH-positive acute respondersare positive responders to long-term treatment withcalcium channel blockers, with almost completenormalization of pulmonary pressures in the controlRHC.
The usefulness of vasoreactivity testing in patientswith heritable PAH, CTD or HIV is not as clear.Nevertheless, current recommendations are toperform the test and seek a long-term response tocalcium channel blockers in those in whom the testis positive.
EVALUATION OF PULMONARY ARTERYHYPERTENSION PROGNOSIS
The severity of PAH is evaluated in patientsafter the diagnostic process and beforetherapeutic decision-making. The clinicalassessment of the patient plays a key role in thechoice of initial treatment, the evaluation of the response, and the possible intensification oftherapy, if required.
The prognostic factors31 used in PAH are basedon patient cohorts and may not accurately reflectthe prognosis of each individual. Scientific evidenceis insufficient to be able to establish the optimalvalues the different parameters should reach or therelative importance of each one. Table 7 summarizesthe most frequently used prognostic factors1,4,21,28 and proposes optimal values for each parameteras an initial framework, although each pulmonaryhypertension center should adapt this according totheir experience and the availability of the differenttests.

Clinical Profile
Age, comorbidities, and the etiology of PAH,together with the presence of heart failure and speedof progression, define how aggressive the disease is.Hemoptysis and atrial arrhythmias (fibrillation andatrial flutter) are typical of severe PAH and indicatepoor prognosis.
Exercise Capacity
This is one of the most robust and classicparameters.32 The 6-minute walking test (6MWT)is the standard test, but with increasing numbers ofpatients in functional class II and with a 6MWT of >400 m, better tools are needed to assess changesin exercise capacity. Cardiopulmonary exercisetesting is the method of choice. Information onthe prognostic value of cardiopulmonary exercisetesting remains limited, but is a field expected toyield important developments in the very nearfuture.33
Biomarkers
N-terminal prohormone brain natriuretic peptideand brain natriuretic peptide are the most widely usedbiomarkers, and are considered excellent markers ofthe severity of RV dysfunction. Their concentrationsincrease when there is progressive deterioration ofexercise capacity or functional class, and decreasewhen there is a positive response to treatment.34
Right Ventricle Function
Right ventricular function is the main determinantof prognosis in patients with PAH.35 The echocardiogram is a simple and readily availabletool to study the following: right chamber dilatation,LV diastolic eccentricity index, tricuspid annularplane systolic excursion,36 TEI index (a combinedmyocardial performance index) and the presenceof pericardial effusion which aid in assessing RVadaptation to pulmonary hypertension. It shouldbe pointed out that estimated pulmonary systolicpressure provides null prognostic information onfollow-up of these patients. Magnetic resonanceimaging25 provides a better assessment of RVfunction: volume, degree of hypertrophy and ejectionfraction. The use of this technique for prognosticstratification of patients with PAH is still in its initialphases, but is of undoubted potential. Finally, RVhemodynamic parameters have been widely used1,4,28 and have a clear role in the assessment of PAHseverity and prognosis.
CLINICAL PATHWAY OF THE PATIENT WITHPULMONARY HYPERTENSION
Pulmonary hypertension, especially PAH andCTEPH, is a serious clinical situation. Diagnosis isusually established in advanced phases of the disease and various specialists are involved in its clinicalmanagement. Similarly, since some of the diagnosticand therapeutic procedures used are highly complexand require experienced professionals to performthem, it is recommended that patients with PAH orCTEPH should be referred to specialized centers.1,4,28 Thus, a clinical pathway is needed to improve patientmanagement.
Typically, suspicion of PH is diagnosed byspecialists in the center nearest to the patient(local hospital). If the initial examinations lead tosuspicion of PAH, CTEPH, or PH of multifactorialorigin, patients should be referred for RHC at a PHcenter (Table 8). Right heart catheterization is notrecommended before referral, except by agreementwith the referral center.1,28 Patients with heart orrespiratory disease with suspected out-of-proportionPH should also be referred to a PH center.

Patients with PH can deteriorate rapidly,28 and so it is important to establish a short timeframe inwhich to complete the diagnostic study and begintreatment; flexible coordination between the localhospital and reference center is essential (Table 9).

If patients live a long way from the referencecenter, they should also be attended by a specialist,preferably with an interest in PH, from a nearbyhospital that would act as local care unit, performfollow-up and provide immediate treatment in caseof complications. In these circumstances, the patientshould follow a care plan agreed between the localhospital and the reference center.
In general, the clinical course of PH is one ofprogressive deterioration with occasional episodesof decompensation. Clinical follow-up should bemaintained until death occurs or transplantation isperformed. In the advanced and irreversible phasesof the disease, palliative care should be mutuallyagreed between the patient, family, local hospitaland the reference center.
Reference Centers
Recently, the Spanish Society of Pneumologyand Thoracic Surgery and the Spanish Societyof Cardiology, in a consensus document at thenational level,28 and the European Society ofCardiology and the European Respiratory Society28 in their international clinical guidelines, maderecommendations regarding the facilities andvolume of activity of the referral center specializedin PH. These are summarized in Table 10.

In particular circumstances, patients with PHrequire highly specialized procedures that may notform part of the functions of the referral center.These situations arise specifically in CTEPH(pulmonary endarterectomy), congenital heartdisease, hereditary PAH, portal hypertension,and lung or heart and lung transplantation. Thereferral center and the specialized centers shouldact in coordination and have preestablished referralprotocols to offer appropriate care to these patient.
CONCLUSIONS
Recently, significant progress has been madein understanding the pathobiology, diagnosis,epidemiology, and prognosis of patients with PAHwhich has led to notable improvements in the qualityand efficacy of clinical care, although a cure has yetto be discovered. Correct diagnosis and an estimateof prognosis using multifactorial approaches are keyaspects of this process and require experience andtechnical training to achieve an optimal result. Thus,a care program and a clinical pathway is warrantedfor patients with PAH.
ABBREVIATIONS
CTEPH: chronic thromboembolic pulmonaryhypertension
HIV: human immunodeficiency virus
IPAH: idiopathic pulmonary arterial hypertension
PAH: pulmonary arterial hypertension
PH: pulmonary hypertension
PWP: pulmonary wedge pressure
RHC: right heart catheterization
Dr Pilar Escribano Subias is a member of the research networkcooperative (REDINSCOR) of the Ministry of Health and Consumers.
Correspondence: Dra. P. Escribano Subias.
Avda. Córdoba, s/n. 28041 Madrid. Spain.
E-mail: pilar.escribano@telefonica.net