

Cardiac amyloidosis is an infiltrative disorder caused by extracellular protein deposition. Transthyretin is a proamyloidotic protein that produces one of the most frequent forms of cardiac amyloidosis, either through mutations or a wild-type form (previously known as senile amyloidosis). Until very recently, diagnosis of transthyretin amyloidosis (ATTR) was very uncommon and histological confirmation was mandatory, making diagnosis of ATTR a real challenge in daily clinical practice. Moreover, the specific therapeutic options to alter the clinical course of the disease were very limited. However, advances in cardiac imaging and diagnostic strategies have improved recognition of ATTR. In addition, several compounds able to modify the natural history of the disease are in the final phases of research, with promising results. Given that effective therapies are on the horizon, cardiologists should be well-versed in this disease and be familiar with its diagnosis and treatment. This review describes the broad clinical spectrum of ATTR in detail, as well as recent advances in the diagnosis and treatment of this condition.
Keywords
Amyloidosis is a deposition disease caused by extracellular accumulation of fibrils whose source consists of proteins with an unstable structure that fold, aggregate, and undergo deposition.1 Such deposition can alter tissue structure and impair the function of various organs and systems.2
Amyloid fibrils are insoluble and proteolysis resistant and are typically stained by Congo red, showing intense yellow-green birefringence under polarized light.3 More than 30 proteins can cause amyloid deposition, but only 5 cause significant deposition in cardiac tissue1:
- •
Light chains, which cause primary amyloidosis (AL).
- •
Transthyretin (TTR), which causes TTR amyloidosis (ATTR).
- •
Apolipoprotein A.
- •
Fibrinogen.
- •
Serum amyloid-protein A, which produces secondary amyloidosis.
Primary amyloidosis and ATTR are the most common forms of cardiac amyloidosis, the AL form being historically considered more common in developed countries.3
Most of the information on cardiac amyloidosis has been based on AL. However, although the number of patients with AL has remained stable, the number ATTR diagnoses has recently increased and it is now thought that ATTR may be much more prevalent than AL.2
Transthyretin amyloidosis has very often been the subject of misdiagnosis or significant delays until its correct diagnosis. Reasons include heterogeneity in its forms, need for histological confirmation, shortages of specialized equipment, and erroneous beliefs among some cardiologists that it is a rare disease without treatment options.2,3
However, these aspects are changing. Diagnosis has implications for patient management. Specific therapies have been developed that may delay or stabilize deposition and that are more effective in the early stages. Early diagnosis is therefore crucial. This review describes significant recent advances in the diagnosis and treatment of ATTR, offering hope for patients with this condition.
TRANSTHYRETIN CARDIAC AMYLOIDOSISTransthyretin is a tetrameric plasma protein responsible for transporting thyroxine and retinol-bound protein. It is primarily synthesized in the liver and secondarily in the choroid plexus and retinal pigment epithelium.4
Transthyretin tends to dissociate to dimers and monomers, which misassemble into fibrils and undergo deposition. Point mutations or the effect of age can increase this tendency, giving rise to the 2 clinical forms of ATTR: mutant (ATTRm) and wild-type (ATTRwt).
MUTANT TRANSTHYRETIN AMYLOIDOSISMore than 120 mutations are currently known to cause ATTRm. These mutations exhibit an autosomal dominant inheritance pattern, with variable penetrance.4 Because of its wide geographic diversity, it is difficult to establish the prevalence of ATTR, but it is considered to be a rare disease with a prevalence of less than 1/100 000 inhabitants2 (Table 1).
Main Clinical and Diagnostic Characteristics of Mutant and Wild-Type Transthyretin Cardiac Amyloidosis
| ATTRwt | ATTRm | |
|---|---|---|
| Prevalence | Unknown. Apparently very frequent | < 1:100 000 |
| Genetic study | Absence of mutations in TTR | Mutation in TTR |
| Typical age at presentation | > 60 y | Variable according to causative mutation |
| Sex | Male predominance. 80% patients | Male predominance, with more aggressive phenotype |
| Extracardiac manifestations | • Carpal tunnel syndrome (33%-49%) • Lumbar spinal stenosis • Traumatic biceps tendon rupture (32%) | • Ascending bilateral sensory-motor polyneuropathy • Dysautonomia: orthostatic hypotension, diarrhea-constipation, erectile dysfunction • Eye involvement: glaucoma, intravitreal deposition, scalloped pupils |
| Cardiac impairment | Constant | Variable according to causative mutation |
| Cardiac output | • Heart failure (53%-86%) • Conduction disorders • AF (43%-67%) • Degenerative AoS | • Conduction disorders • Heart failure • Infrequent AF (10%) |
| Diagnostic techniques | ||
| ECG | • Pseudoinfarct pattern (63%-66%) • Low voltage (22%-33%) • Sokolow LVH (6%-13%) | • Pseudoinfarct pattern (18%-69%) • Low voltage (2%-25%) • Sokolow LVH (3%-8%) |
| ECHO | • Moderate-severe hypertrophy • Mild-moderate depressed LVEF (30%) | • Moderate hypertrophy • LVEF, typically preserved |
| Cardiac MRI | • Late enhancement • Elevated native T1 and EV | |
| 99mTc DPD scintigraphy | • Grade 2-3 | • Grade 0: asymptomatic carriers • Grade 1: initial cardiac involvement • Grade 2-3: significant cardiac involvement |
AF, atrial fibrillation; AoS, aortic stenosis; ATTRm, mutant transthyretin amyloidosis; ATTRwt, wild-type transthyretin amyloidosis; ECG, electrocardiogram; ECO, echocardiogram; EV, extracellular volume; LVEF, left ventricular ejection fraction; LVH, left ventricular hypertrophy; TTR, transthyretin.
The first TTR mutations were reported as familial amyloid polyneuropathy (or Andrade disease), and consequently ATTRm has until recently been considered a neurologic disease. However, recent findings show cardiac involvement in more than half of the cases.3
There is a strong genotype-phenotype correlation, with mutations being associated with purely neurologic disease or purely cardiac disease.3 However, the division of ATTRm into cardiac or neurologic disease may be an oversimplification, as there is considerable overlap between the 2 clinical forms on the disease spectrum.
The Val30Met mutation (now known as Val50Met after 20 positions were added to the traditional mutation name in ATTRm) is the most frequent mutation worldwide and is endemic in Portugal, Japan, and Sweden. Its estimated incidence in Portugal is 1 per 538 inhabitants.2 Mallorca (Spain) and Valverde del Camino (Huelva, Spain) are also considered to be areas in which ATTRm is endemic. The estimated prevalence in Mallorca in symptomatic patients is 3/100 000 inhabitants.5
The Val30Met mutation causes a predominantly neurologic condition with symmetric sensory-motor polyneuropathy, which begins in the lower limbs and follows an ascending pattern. It can be associated with dysautonomia with orthostatic hypotension, erectile dysfunction, urinary incontinence, and gastrointestinal symptoms. It typically begins at the end of the second or third decade of life, and up to 43% of the patients have cardiac involvement that is a frequent cause of death4 (Table 1).
Of particular relevance is the Val122Ile mutation (p. Val142Ile), which is present in 3% to 4% of the North American black population.3 Although its penetrance is incomplete,3 this mutation has been associated with a 47% increased risk of developing heart failure (HF).6 A recent study showed that Val122Ile amyloidosis was the fourth most common cause of HF in the British Afro-Caribbean population.7 Although up to 30% of patients with this mutation may have features of mild neuropathy,6 the clinical phenotype is usually similar to that of ATTRwt.4 Val122Ile should not be considered a mutation exclusive to the black population, because it can also be present in the white population. For example, we have identified this mutation in 4 white Spanish families without black ancestry.
WILD-TYPE TRANSTHYRETIN AMYLOIDOSISWild-type transthyretin amyloidosis was first described in 1876. It was formerly called senile amyloidosis, but its diagnosis in patients aged 40 to 60 years has rendered this term obsolete. Of interest, the earliest known case of this mutation was found in a 47-year-old American patient.8
The exact prevalence of ATTRwt remains unknown. However, studies suggest that it is underdiagnosed and that it may be the most frequent form of cardiac amyloidosis.2,3 The following results support this hypothesis:
- •
In patients aged more than 80 years, the prevalence of TTR deposition is 25% at autopsy.3
- •
In patients with HF with preserved ejection fraction (HFpEF), moderate-severe TTR deposition is 5% at autopsy.9
- •
In patients aged more than 60 years admitted for HFpEF and left ventricular hypertrophy (LVH) ≥ 12mm, our group recently found a prevalence of 13%.10
Unlike ATTRm, ATTRwt is a sporadic disease that typically begins after age 70 years.4 It is mainly found in men, and published series have reported rates ranging from 89% to 98%.11,12 However, in a recent series of patients diagnosed with ATTRwt in 2 hospitals (Madrid, Spain and Bologna, Italy), our group found that 20% were women. Furthermore, other autopsy studies have also suggested that ATTRwt in women may be more widespread than previously reported. Therefore female sex should not decrease clinical suspicion of ATTRwt (Table 1).13
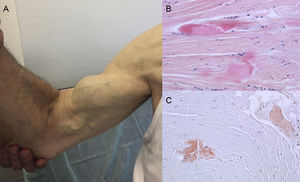
Autopsy findings show that TTR deposition is dispersed in different organs in ATTRwt. However, deposition is much greater in the heart due to the cardiac tropism of TTR, and cardiac involvement is the main clinical manifestation.4 Patients may present with symptoms of extracardiac TTR deposition such as lumbar canal stenosis, atraumatic rupture of the biceps tendon or “Popeye sign”, and carpal tunnel syndrome (CTS)3 (Figure 1). All these features may help to guide and promptly establish the diagnosis. CTS may accompany other amyloidosis subtypes, but it is more common in ATTRwt. Deposition may precede cardiac manifestations by several years.6 It may be used as an indication in elderly patients with LVH, especially if they have bilateral CTS not associated with specific occupational activities and are in New York Heart Association functional class ≥ II (unpublished data).
. B and C: staining with hematoxylin-eosin (B) and Congo red (C), both ×200, of carpal ligament sample showing dense collagen bundles with noncellular material. Courtesy of Dr Clara Salas Antón.")
Signs and symptoms of transthyretin amyloidosis. A: nontraumatic rupture of the right biceps tendon (“Popeye sign”). B and C: staining with hematoxylin-eosin (B) and Congo red (C), both ×200, of carpal ligament sample showing dense collagen bundles with noncellular material. Courtesy of Dr Clara Salas Antón.
Amyloid can infiltrate any cardiac structure.1 Typically, deposition increases ventricular wall thickness, which causes a gradual decrease in distensibility leading to severe diastolic dysfunction. ATTR has therefore traditionally been included as a cause of restrictive cardiomyopathy.
However, the clinical spectrum of ATTR is much broader and more heterogeneous. The most common symptom of ATTR is HF. As mentioned, a study published by our group in 2015 suggested that a protocol based on 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid (99mTc-DPD) scintigraphy may be useful for the diagnosis of ATTRwt in a significant proportion (13%) of patients older than 60 years admitted for HFpEF.10 Based on this result, 99mTc-DPD scintigraphy was included in the 2016 European guidelines on HF as a useful tool for the identification of patients with ATTR.14 However, ATTR should not be suspected exclusively in patients with HFpEF because, as amyloid deposition advances the contractile function worsens, and consequently ATTR can be associated with different degrees of systolic dysfunction.
Transthyretin amyloidosis is a phenocopy of hypertrophic cardiomyopathy (HCM) and may be confused with it. A recent multicenter French study reported that 5% of patients with HCM have ATTRm.15 However, our results are not in line with this high rate, which could be related to the large black population in France.
Cardiac conduction abnormalities may be the first manifestation of ATTR. Amyloid infiltration of the sinus and atrioventricular nodes1 may indicate the need for pacemaker implantation (Table 1). The previously mentioned study conducted in Spain and Italy found that conduction disorders were the first manifestation of ATTRwt in 7% of patients with this disease.13
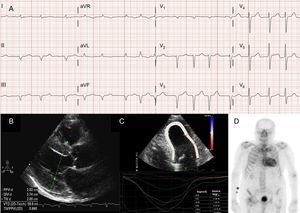
Atrial arrhythmias are also very common in patients with ATTRwt13 (Figure 2A), and the first manifestation of the disease can be stroke.4 In fact, the Mayo Clinic group recently suggested that ATTRwt should be ruled out in the setting of a diagnosis of non-valvular atrial fibrillation (AF) in elderly patients.8 In contrast, AF is much less common patients with ATTRm (Table 1).
. A: electrocardiogram of a patient with wild-type transthyretin amyloidosis (ATTRwt), showing atrial fibrillation and pseudoinfarct pattern in inferior leads. B: echocardiogram of a patient with mutant transthyretin amyloidosis with Val30Met mutation, with marked concentric left ventricular hypertrophy and mild pericardial effusion. C: longitudinal regional strain of patient with ATTRwt, showing preserved values in the apical segment and depressed values in the basal and midventricular segments. D, 99mTc-DPD (99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid) scan of a patient with ATTRwt, showing biventricular uptake superior to bone uptake, corresponding to Perugini grade 3.")
Diagnostic techniques in transthyretin cardiac amyloidosis (ATTR). A: electrocardiogram of a patient with wild-type transthyretin amyloidosis (ATTRwt), showing atrial fibrillation and pseudoinfarct pattern in inferior leads. B: echocardiogram of a patient with mutant transthyretin amyloidosis with Val30Met mutation, with marked concentric left ventricular hypertrophy and mild pericardial effusion. C: longitudinal regional strain of patient with ATTRwt, showing preserved values in the apical segment and depressed values in the basal and midventricular segments. D, 99mTc-DPD (99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid) scan of a patient with ATTRwt, showing biventricular uptake superior to bone uptake, corresponding to Perugini grade 3.
Finally, we note that ATTR and degenerative aortic stenosis can coexist in the same patient. In 2016, several studies drew attention to this possibility, and a prospective study reported that ATTRwt had a prevalence of 6% in patients older than 65 years who underwent aortic valve replacement.16 This study suggested that patients with both entities had a much worse postoperative prognosis than those without ATTRwt (mortality 50% vs 6.9% after a median follow-up of 2.3 years).16 Another recent study with 99mTc-DPD scintigraphy in 43 patients with low-flow/low-gradient aortic stenosis identified 5 patients with ATTRwt (prevalence 12%). 17 Patients with severe aortic stenosis and ATTRwt share the same demographic profile, and the appropriate treatment for patients with both diseases is still to be determined.
Usefulness of Diagnostic TechniquesThe diagnosis of ATTR is a challenge in daily clinical practice. Although electrocardiography and echocardiography play a role in diagnosis, new noninvasive techniques have acquired a key role in the assessment of patients with ATTR.
ElectrocardiogramThe association between low voltage and cardiac amyloidosis has long been considered indisputable.3 The most widely used criteria in clinical practice are QRS amplitude < 1 mV in all precordial leads or < 0.5 mV in all limb leads.1 Although low electrocardiographic voltages in the setting of LVH should establish suspicion, prevalence in a contemporary series of ATTR was as low as 20% to 25%.3,4,13 Prevalence also varies according to the criteria applied. For example, use of the Sokolow criterion (S wave in lead V1 + R wave in lead V5 or V6<1.5 mV) can increase calculated prevalence to between 46% and 58%.13 The ratio of left ventricular wall thickness to total QRS voltage has been recommended to better assess disparities between the results of the 2 techniques.2,3 However, up to 20% of patients with ATTR can meet electrocardiographic criteria for LVH.2,3
In most series of patients with cardiac amyloidosis, the pseudoinfarct pattern is the most common electrocardiographic finding2,3,13 (Figure 2A). Due to possible conduction system involvement, complete or incomplete bundle branch blocks are also common.3
EchocardiographyAlthough echocardiography is the cornerstone of the initial diagnosis of ATTR, no findings are specific.3 Transthyretin amyloidosis has typically been associated with a normal or small left ventricle with concentric hypertrophy.3 The 10th International Symposium on Amyloid and Amyloidosis held in 2004 established the echocardiographic criterion of heart disease due to AL in the absence of other causes of LVH as the presence of LVH with a cutoff of 12 mm for interventricular septal wall thickness.4 This criterion was later extrapolated to other forms of amyloidosis (Figure 2B), which conferred a high degree of specificity but low sensitivity.
Although concentric LVH has been classically described, current series suggest that about 20% have asymmetric LVH.13
Despite the classic association between a normal or slightly decreased left ventricular ejection fraction (LVEF) and cardiac amyloidosis,2 the LVEF range is highly variable.8 In a recent study conducted at the Mayo Clinic, an LVEF < 50% was observed in almost half of the patients with ATTRwt,8 whereas in our series an LVEF < 50% was observed in 37% of patients.13 In addition, the use of LVEF in the assessment of systolic function in cardiac amyloidosis is limited, because slightly depressed values are already indicative of relevant cardiac disease. This limitation can be overcome by the use of tissue Doppler velocities, strain imaging, and myocardial contraction fraction, which have been proposed as more appropriate indices to assess cardiac function.2
Other classic echocardiographic signs are right ventricular hypertrophy, biatrial dilatation, mild pericardial effusion, atrioventricular valve thickening, atrial septal wall thickening, and granular sparkling appearance of the myocardium.3,6 However, because some of these features were observed in a highly selected series of patients in advanced stages of disease, not all of them have to be present to establish suspicion.1
Regional strain imaging is a very useful technique for the early diagnosis of patients with ATTR. In patients with ATTR, longitudinal strain is depressed in basal and midventricular segments but is preserved in apical segments18 (Figure 2C). This typical pattern can be useful in the differential diagnosis of ATTR from other heart diseases.4
BiomarkersThere are fewer data on the role of the N-terminal prohormone of brain natriuretic propeptide (NT-proBNP) and troponin in ATTR than there is in AL.4 The NT-proBNP levels in ATTR are typically lower than in AL,4 reflecting 2 different pathophysiological mechanisms: direct light-chain toxicity in AL vs induced tissue damage by protofibrils in ATTR.
Recently, the Mayo Clinic group proposed a stratification system similar to that in force for AL. In a cohort of 360 patients with ATTRwt, both biomarkers were shown to be predictors of mortality. Stage III patients (NT-proBNP > 3000 pg/mL and troponin T > 0.05 ng/mL) had a median survival of 20 months, whereas stage I and II patients had a median survival of 66 months and 40 months (no biomarker or just 1 biomarker above the established cutoff points, respectively).
Cardiac Magnetic Resonance ImagingCardiac magnetic resonance imaging (CMRI) can be used to obtain structural and functional information and characterize the composition of myocardial tissue.3 The CMRI is essential in the early identification of ATTR, and in its differential diagnosis from other heart disease.
The characterization of tissue by CMRI is based on the following features:
- •
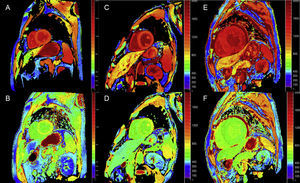
Late enhancement: A global subendocardial pattern is virtually pathognomonic of cardiac amyloidosis, but is only present in about a quarter of patients. Other patterns, such as transmural (the most common) or patching, are also compatible (Figure 3). Despite its high sensitivity and specificity, it should be taken into account that there may be a possible absence of late enhancement (15% of patients) and, in our experience, a nonnegligible percentage of false negatives for technical reasons.3 The transmural enhancement pattern is associated with worse prognosis and is an independent predictor of mortality.19
, showing diffuse pathological transmural gadolinium deposition. C and D: late enhancement sequences, 4-chamber and short-axis basal level, respectively, of patients with ATTRm showing pathological gadolinium deposition with a patched pattern, with lower inferoseptal and inferolateral basal focal area. E and F, late enhancement sequences, 4-chamber plane and short axis at the apical level, respectively, of patients with ATTRm, showing extensive pathological transmural deposition, except in basal and middle anterolateral segments. Courtesy of Dr Jesús González Mirelis.") Figure 3.
Figure 3.Diversity of late enhancement patterns by cardiac magnetic resonance imaging in transthyretin amyloidosis. A and B: late enhancement sequences, 4-chamber plane and short axis at the mid level, respectively, of a patient with mutant transthyretin amyloidosis (ATTRm), showing diffuse pathological transmural gadolinium deposition. C and D: late enhancement sequences, 4-chamber and short-axis basal level, respectively, of patients with ATTRm showing pathological gadolinium deposition with a patched pattern, with lower inferoseptal and inferolateral basal focal area. E and F, late enhancement sequences, 4-chamber plane and short axis at the apical level, respectively, of patients with ATTRm, showing extensive pathological transmural deposition, except in basal and middle anterolateral segments. Courtesy of Dr Jesús González Mirelis.
(0.15MB). - •
Long T1 times: T1 mapping is a technique in which a quantitative myocardial signal is measured before (native T1) or after contrast administration. Native T1 times are very long in cardiac amyloidosis.3 T1 mapping does not require the administration of contrast and so can be used in kidney failure. T1 times may even be abnormal before LVH is observed.3 T1 times are longer in ATTR than in HCM and controls (1097ms ± 43 ms vs 1026ms ± 64 ms vs 9.67ms ± 34ms, respectively; P < .0001), but are shorter than those in AL (1130ms ± 68 ms; P = 0.01).20
- •
Contrast administration can be used to calculate extracellular volume (ECV) and assess increases of extracellular space. ECV values in cardiac amyloidosis are higher than in other heart disease, except in myocardial infarct zones.21 In 2016, our group, in collaboration with other national centers, reported that ECV quantification can identify cardiac involvement in ATTRm and, for the first time, correlated it with the degree of neurologic impairment, supporting the use of this technique in early diagnosis and tracking of ATTRm.22
, showing diffuse pathological transmural gadolinium deposition. C and D: late enhancement sequences, 4-chamber and short-axis basal level, respectively, of patients with ATTRm showing pathological gadolinium deposition with a patched pattern, with lower inferoseptal and inferolateral basal focal area. E and F, late enhancement sequences, 4-chamber plane and short axis at the apical level, respectively, of patients with ATTRm, showing extensive pathological transmural deposition, except in basal and middle anterolateral segments. Courtesy of Dr Jesús González Mirelis.")
Quantitative T1 mapping and ECV calculation techniques can be very useful in ATTR for early diagnosis, clinical follow-up, and treatment response assessment (Figure 4).
 in 3T cardiac magnetic resonance imaging in healthy control, patient with transthyretin amyloidosis, and patient with primary light-chain amyloidosis. A and B: native T1 mapping and extracellular volume (EV), respectively, in a healthy control, showing normal values (EV = 0.214). C and D: native T1 mapping and EV, respectively, in a patient with mutant transthyretin amyloidosis with neurologic damage and incipient cardiac involvement, elevated native T1, and slightly elevated EV (0.361). E and F: native T1 mapping and EV, respectively, in a patient with wild-type transthyretin cardiac amyloidosis, elevated native T1, and very high EV (0.626), reflecting massive amyloid infiltration. Courtesy of Dr Jesús González Mirelis.")
T1 mapping, before and after contrast, with modified look-locker inversion-recovery (MOLLI) in 3T cardiac magnetic resonance imaging in healthy control, patient with transthyretin amyloidosis, and patient with primary light-chain amyloidosis. A and B: native T1 mapping and extracellular volume (EV), respectively, in a healthy control, showing normal values (EV = 0.214). C and D: native T1 mapping and EV, respectively, in a patient with mutant transthyretin amyloidosis with neurologic damage and incipient cardiac involvement, elevated native T1, and slightly elevated EV (0.361). E and F: native T1 mapping and EV, respectively, in a patient with wild-type transthyretin cardiac amyloidosis, elevated native T1, and very high EV (0.626), reflecting massive amyloid infiltration. Courtesy of Dr Jesús González Mirelis.
In the 1980s, observation of cardiac uptake of several bone diphosphonate tracers was histologically correlated to the presence of cardiac amyloidosis.23 The mechanism of uptake is not well characterized, but it may be related to the calcium content of amyloid deposits.
An early study by the Bologna group using 99mTc-DPD found cardiac uptake in 15 patients with ATTR and its absence in 10 patients with AL, using a score based on biventricular uptake equal or superior to bone uptake (Perugini score)24 (Figure 2D). Similar findings were subsequently reported by our group and others.25 Mild uptake (score 1) and moderate uptake (score 2) can be found in 30% and 10% of patients with AL, respectively.24
Given its high sensitivity and specificity, this technique is extremely useful for establishing a diagnosis of ATTR and may show cardiac involvement even when echocardiography and MRI findings are still normal. In fact, after scintigraphy for oncologic or rheumatologic indications, incidental findings of ATTR are not uncommon.26
The Tc-DPD is not available in the United States, but similar results have been reported using 99mTc-PYP (pyrophosphate) imaging.27
Other radiotracers are currently under study. For example, 18F-florbetapir, which has already been approved for brain beta-amyloid imaging,4 has been studied in patients with AL and ATTR. The results show that 18F-florbetapir can detect myocardial AL and ATTR deposits.28 Although the available data were obtained in case studies29 and the high cost of this radiotracer limits its use, several studies are underway on the potential advantage of its use over that of Tc-DPD as a screening technique for the 2 most common types of amyloidosis.
Invasive DiagnosisThe definitive diagnosis of ATTR is based on the histological demonstration of amyloid fibrils. Although there may be extracardiac deposition, the likelihood of demonstrating amyloid by histology varies by organ.2 There are few studies on the cost-effectiveness of extracardiac biopsy (eg, abdominal fat, gingival, salivary gland, gastrointestinal) in ATTR, which is greater in ATTRm than in ATTRwt. However, a negative biopsy of a clinically unaffected organ does not exclude a diagnosis of ATTR.4
As in ATTRwt, endomyocardial biopsy is indicated in patients with no extracardiac involvement or with heart disease alone.3,4 Endomyocardial biopsy is a low-risk procedure (especially in experienced centers) and sampling errors are unlikely.6
After histological confirmation of amyloidosis, which may sometimes require interpretation by trained personnel,6 correct classification of the subtype is crucial.4 Currently, classification depends on a combination of immunohistochemistry, genetic analysis, and proteomics:
- •
Immunohistochemistry is based on the use of specific antibodies against known amyloid proteins. Although the results of this technique are typically definitive, it is less sensitive in the recognition of light chains.4
- •
This limitation can be overcome by the use of mass spectrometry, which provides definitive results and is the criterion standard in the confirmation of the amyloid subtype.2 Although this technique is only available in specialist centers, it is especially useful in inconclusive cases or in cases that are positive for several antibodies on immunohistochemistry, which in our experience occurs in about 20% to 30% of cases. 4
- •
Because clinical or histological techniques cannot distinguish ATTRm from ATTRwt, genetic studies are recommended in all cases of ATTR. The finding of a causative mutation can be of importance for offering genetic counseling and follow-up to asymptomatic carriers, 4,30 who could benefit from forthcoming therapies that delay or even prevent the onset of the disease.31
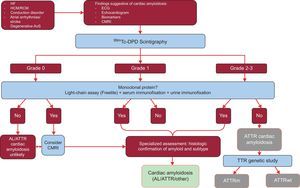
Until recently, histological studies were considered essential in the diagnosis of ATTR.3 However, to facilitate diagnosis, in 2016 an international multicenter study proposed a new algorithm for the noninvasive diagnosis of ATTR.32
The study analyzed the results of 1217 patients. The presence of classic signs of cardiac amyloidosis using imaging techniques, grade 2 or 3 Tc-DPD/PYP uptake on scintigraphy, and the absence of a monoclonal protein had a specificity and positive predictive value for ATTR of 100%32 (Figure 5).

Diagnostic algorithm for patients with suspected cardiac amyloidosis. 99mTc-DPD scintigraphy grading system: grade 0, no cardiac uptake; grade 1, slight lower uptake than bone; grade 2, moderate uptake equal to bone uptake; grade 3, severe uptake superior to bone uptake. ACV, stroke; AL, primary light-chain amyloidosis; AoS, aortic stenosis; ATTR, transthyretin amyloidosis; ATTRm, mutant transthyretin amyloidosis; ATTRwt, wild-type transthyretin amyloidosis; CMRI, cardiac magnetic resonance imaging; ECG, electrocardiogram; HCM, hypertrophic cardiomyopathy; HF, heart failure; RCM, restrictive cardiomyopathy; TTR, transthyretin.
A key feature this algorithm is the absence of a monoclonal protein that could cause AL on serum chain assay (Freelite, The Binding Site, UK) and on immunofixation electrophoresis of blood and urine. The presence of a monoclonal protein is an indication for endomyocardial biopsy to distinguish between ATTR and AL.32 Up to 5% of the population aged more than 65 years have monoclonal gammopathy of undetermined significance.2 In elderly individuals, a moderate increase in circulating light chains should not directly lead to a diagnosis of AL. It has been reported that up to 10% of elderly patients with ATTRwt and monoclonal gammopathy of undetermined significance in reference centers had previously received a misdiagnosis of AL.3,33 A correct diagnosis is needed to avoid inappropriate chemotherapy. Of interest, our hospital has documented 2 cases of patients with multiple myeloma and concomitant ATTRwt on mass spectrometry.
TREATMENT OF TRANSTHYRETIN CARDIAC AMYLOIDOSISTreatment of patients with ATTR has 2 objectives: to provide medical support and, if possible, to stop or delay amyloid deposition by the use of specific treatments.
Medical TreatmentThe following sections describe supportive cardiac care for patients with ATTR.
Management of Heart FailureEuvolemia must be maintained in patients with cardiac amyloidosis. Diet and lifestyle measures are very important. Diuretics are key to the treatment of HF in ATTR. However, because excessive use of diuretics can lead to hypotension (frequently due to autonomic dysfunction) and worsen the clinical situation, especially in ATTRm, extreme care must be exercised in its management.
In the treatment of HF in ATTR, it must be taken into account that impaired diastolic dysfunction and reduced stroke volume lead to compensatory tachycardia to maintain cardiac output. Therefore, beta-blockers must be used with care and individualized for each patient. Standard practice is to remove them in the absence of difficulties in controlling heart rate. This approach is even more important, if possible, in ATTRwt because of the frequent presence of conduction disorders.6 Calcium antagonists and digoxin can bind to amyloid fibrils and are therefore contraindicated in ATTR owing to the risk of toxicity even at therapeutic doses.6
In contrast to HF with systolic dysfunction due to other etiologies, there is no evidence in support of a prognostic benefit due to the use of beta-blockers, angiotensin-converting enzyme inhibitors, or angiotensin II receptor antagonists in cardiac amyloidosis. In fact, their use may lead to clinical worsening due to hypotension and low output: a recent publication has reported worse prognosis in ATTRm and a neutral effect in ATTRwt.34
Management of Atrial ArrhythmiasThe management of AF in ATTR is a challenge. Maintaining long-term sinus rhythm is difficult. However, electrical cardioversion may be attempted because it can lead to clinical improvement.
Thromboembolic risk in patients with ATTR is very high. In addition, chronic amyloid infiltration can lead to mechanical atrial dysfunction, which may be the underlying cause of the development of atrial thrombus in some patients without AF. Anticoagulant therapy in ATTR should not be based on the CHADS2-VASC score and should be standard therapy in AF. Bleeding events are less common than in AL, and so some hospitals recommend anticoagulant therapy in sinus rhythm patients if there is poor atrial function according to transmitral Doppler velocities. Although there are no comparative studies on the effectiveness of direct oral anticoagulants vs vitamin K antagonists, our hospital has administered direct oral anticoagulants to selected patients.
Role of DevicesCurrent recommendations for pacemaker implantation are the same in ATTR and the general population. However, some groups favor prophylactic implantation, especially in patients with ATTRm and conduction disorders.35 We do not favor this preventative strategy and have not found such a high rate of conduction disorders to warrant prophylactic implantation in patients with ATTRm.
The role of implantable cardioverter-defibrillator (ICD) use in ATTR is not well established. In a small series, ICD implantation did not significantly improve survival, although it had an appropriate effect in multiple patients during the first 2 years.36
Heart TransplantationHeart transplantation has played a minor role in ATTR because ATTRm can involve various organs and ATTRwt typically affects elderly patients. However, the absence of extracardiac involvement in patients with ATTRwt makes them good candidates for the procedure. The literature provides examples of successful transplantation in patients younger than 70 years with ATTRwt or with ATTRm and predominant cardiac involvement.4
Specific Treatment of Transthyretin Cardiac AmyloidosisAt present, there is no approved therapy for the specific treatment of ATTR cardiac amyloidosis, although liver transplantation (TxH) alone or in combination with heart transplantation has been used in ATTRm since the 1990s as a way to eliminate the main source of precursor TTR.4
Liver TransplantationThe Familial Amyloidotic Polyneuropathy World Transplant Registry37 reported that more than 2000 patients with ATTRm have undergone TxH in 20 countries.4 Patients with the Val30Met mutation and a predominantly neurologic clinical picture have a survival rate of more than 50% at 20 years.3 These promising results are based on strict patient selection according to age, type of mutation, and stage of the disease. The most commonly accepted indication for TxH is the combination of young age, the Val30Met mutation, and early stages of the disease.
However, the main limitations of this technique are the shortage of donors, the need for chronic immunosuppression, advanced age at the time of presentation, and worse results obtained in patients with mutations other than the Val30Met mutation.
In addition, the theoretical suppression of production of the mutated protein is counteracted by postimplantation progressive native TTR deposition,4,6 whose mechanism is not completely understood. In fact, cardiac TTR deposition after TxH affects morbidity and mortality.
The need to better understand the pathogenesis of ATTR and the limitations of TxH has stimulated the development of several drugs.
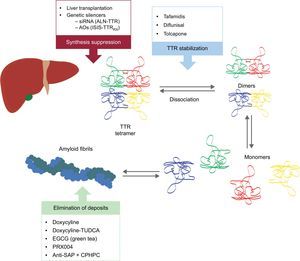
These new compounds act at different points in the TTR amyloidogenesis cascade (Figure 6). Treatment will always involve reducing the precursor protein, although avoiding deposition and eliminating existing depositions will be equally important. Therefore, we believe that, in future, the approach to this disease will be in the form of combined treatment.
![Specific therapies in transthyretin cardiac amyloidosis and main targets. AntiSAP + CPHPC, antiserum amyloid P component + (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid; AOs, antisense oligonucleotides; EGCG, epigallocatechin-3 gallate; siRNA, small interfering RNA; TTR, transthyretin; TUDCA, tauroursodeoxycholic acid.](https://static.elsevier.es/multimedia/18855857/0000007000000011/v1_201710271222/S1885585717303857/v1_201710271222/en/main.assets/gr6.jpeg?xkr=eyJpdiI6IkhuZHIzZEJOQTFmRUo0N0RZNCtabEE9PSIsInZhbHVlIjoiMkNnWWNqVWp5MGxxa0QrZUhrSkJHL2tjV05KcFczYTd3aTlyaEZDckpzdWRyVTF1MVg2OE5tMFlyR0I2YTNzK1llYUEyMEhFL1dnRG15M0FCNGxSZVRUVXZVSGt3V203MFpWeExHeTlONWpkQ3JSUHFpMkR6YytMMU82anliVytWOVFUalhXU3dQcHpLaVZoeHBPbzZXamdwdnRmbjNUVGt6T29tNEo0V3VpMy9qWEtRaGkwWUlhUjYyMkNMUGVCRTlhNEJWeTdFTmVucG1JNHB4K1MvWWRjYWIyQlVQUmZBSzhEQm5YVnRBZXFpL3lHVlFiamZiOVZjc25vYUtqK2pmVGg3OG85YUdFK01TWm4zc3p4V0lhN1poZVZwVzlOcUtYcytEeDJwY0U9IiwibWFjIjoiOGFhODNkODEwN2RkYTYwMTcxZTM2ZjQyZTM3M2RkNGNiOTg4M2QyOGEzMTlkNmUzMTZlYWEyNGVhOTk0ODE4MiIsInRhZyI6IiJ9 "Specific therapies in transthyretin cardiac amyloidosis and main targets. AntiSAP + CPHPC, antiserum amyloid P component + (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid; AOs, antisense oligonucleotides; EGCG, epigallocatechin-3 gallate; siRNA, small interfering RNA; TTR, transthyretin; TUDCA, tauroursodeoxycholic acid.")
Specific therapies in transthyretin cardiac amyloidosis and main targets. AntiSAP + CPHPC, antiserum amyloid P component + (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid; AOs, antisense oligonucleotides; EGCG, epigallocatechin-3 gallate; siRNA, small interfering RNA; TTR, transthyretin; TUDCA, tauroursodeoxycholic acid.
Two research lines are underway on the inhibition of TTR liver expression: the use of small interfering RNA (siRNA) and the use of antisense oligonucleotide (AO) drugs.
- •
The siRNA are double-stranded RNA molecules that silence messenger RNA sequences by specifically binding to them, preventing protein formation. Patisiran (ALN-TTR02) has been found to reduce TTR production by 80%.38 In patients with ATTRm, TTR reduction was 87%.39 A phase 2 trial has shown promising results, demonstrating stable echocardiographic, functional, and analytical parameters at 12 months and 24 months.40 The results of the phase 3 neurologic study in patients with ATTRm and a subanalysis of patients with cardiac involvement are expected in 2017 (Table 2). Another drug, revusiran (ALN-TTR01), is administered subcutaneously and differs from patisiran in the lipid nanoparticles that encapsulate siRNA. This drug was the subject of a phase III clinical trial in patients with ATTRm who have heart disease. The study was discontinued last year due to an unexpected increase in mortality in the treatment group (Table 2).
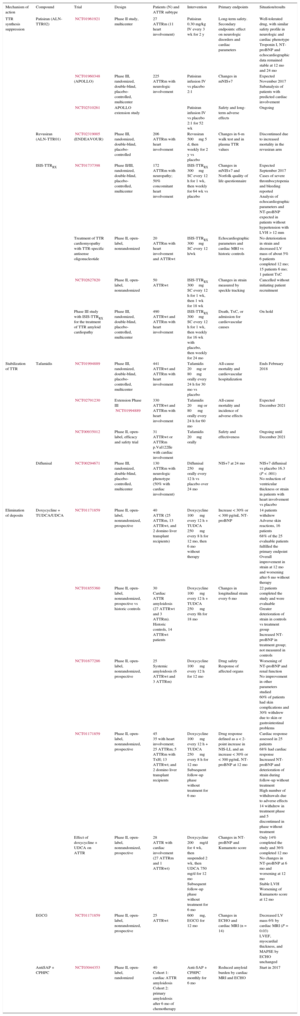
Table 2.Main Ongoing Clinical Trials in Transthyretin Cardiac Amyloidosis
Mechanism of action Compound Trial Design Patients (N) and ATTR subtype Intervention Primary endpoints Situation/results TTR synthesis suppression Patisiran (ALN-TTR02) NCT01961921 Phase II study, multicenter 27
ATTRm (11 heart involvement)Patisiran 0.30 mg/kg IV every 3 wk for 2 y Long-term safety. Secondary endpoints: effect on neurologic disorders and cardiac parameters Well-tolerated drug, with similar safety profile in neurologic and cardiac phenotype
Troponin I, NT-proBNP and echocardiographic data remained stable at 12 mo and 24 moNCT01960348 (APOLLO) Phase III, randomized, double-blind, placebo-controlled, multicenter 225
ATTRm with neurologic involvementPatisiran infusion IV vs placebo 2:1 Changes in mNIS+7 Expected November 2017
Subanalysis of patients with predicted cardiac involvementNCT02510261 APOLLO extension study Patisiran infusion IV vs placebo 2:1 for 52 wk Safety and long-term adverse effects Ongoing Revusiran (ALN-TTR01) NCT02319005 (ENDEAVOUR) Phase III, randomized, double-blind, placebo-controlled 206
ATTRm with heart involvementRevusiran 500mg 5 d, then weekly for 2 y vs placebo Changes in 6-m walk test and in plasma TTR values Discontinued due to increased mortality in the revusiran arm ISIS-TTRRX NCT01737398 Phase II/III, randomized, double-blind, placebo-controlled, multicenter 172
ATTRm with neuropathy; 50% concomitant heart involvementISIS-TTRRX 300mg SC every 12 h for 1 wk, then weekly for 64 wk vs placebo Changes in mNIS+7 and Norfolk quality of life questionnaire Expected September 2017
Cases of severe thrombocytopenia and bleeding reported
Analysis of echocardiographic parameters and NT-proBNP expected in patients without hypertension with LVH > 12 mmTreatment of TTR cardiomyopathy with TTR-specific antisense oligonucleotide Phase II, open-label, nonrandomized 20
ATTRm with heart involvement and ATTRwtISIS-TTRRX 300mg SC every 12 h/wk Echocardiographic parameters and cardiac MRI vs historic controls No deterioration in strain and decreased LV mass of about 5%
6 patients completed 12 mo; 15 patients 6 mo; 1 patient TxCNCT02627820 Phase II, open-label, nonrandomized 50
ATTRwtISIS-TTRRX 300mg SC every 12 h for 1 wk, then 1 wk for 18 wk Changes in strain measured by speckle tracking Cancelled without initiating patient recruitment Phase III study with ISIS-TTRRX for the treatment of TTR amyloid cardiopathy Phase III, randomized, double-blind, placebo-controlled, multicenter 490
ATTRwt and ATTRm with heart involvementISIS-TTRRX 300mg SC every 12 h for 1 wk, then weekly for 16 wk with placebo, then weekly for 24 mo Death, TxC, or admission for cardiovascular causes On hold Stabilization of TTR Tafamidis NCT01994889 Phase III, randomized, double-blind, placebo-controlled, multicenter 441
ATTRwt and ATTRm with heart involvementTafamidis 20mg or 80mg orally every 24 h for 30 mo vs placebo All-cause mortality and cardiovascular hospitalization Ends February 2018 NCT02791230 Extension Phase III NCT01994889 330
ATTRwt and ATTRm with heart involvementTafamidis 20 mg or 80 mg orally every 24 h for 60 mo All-cause mortality and incidence of adverse effects Expected December 2021 NCT00935012 Phase II, open-label, efficacy and safety trial 31
ATTRwt or ATTRm p.Val122Ile with cardiac involvementTafamidis 20mg orally Safety and effectiveness Ongoing until December 2021 Diflunisal NCT00294671 Phase III, randomized, double-blind, placebo-controlled, multicenter 130
ATTRm with neurologic phenotype (50% with cardiac involvement)Diflunisal 250mg orally every 12 h vs placebo over 24 mo NIS+7 at 24 mo NIS+7 diflunisal vs placebo 16.3 (P < .001)
No reduction of ventricular thickness or strain in patients with heart involvement vs placeboElimination of deposits Doxycycline + TUDCA/UDCA NCT01171859 Phase II, open-label, nonrandomized, prospective 40
ATTR (25 ATTRm, 13 ATTRwt, and 2 domino liver transplant recipients)Doxycycline 100 mg every 12 h + TUDCA 250 mg every 8 h for 12 mo, then 6 mo without therapy Increase < 30% or < 300 pg/mL NT-proBNP 14 patients withdrew
Adverse skin reactions, 16 patients
68% of the 25 evaluable patients fulfilled the primary endpoint
Overall improvement in strain at 12 mo and worsening after 6 mo without therapyNCT01855360 Phase II, open-label, nonrandomized, prospective vs historic controls 30
Cardiac ATTR amyloidosis (27 ATTRwt and 3 ATTRm). Historic controls, 14 ATTRwt patientsDoxycycline 100 mg every 12 h + TUDCA 250 mg every 8h for 18 mo Changes in longitudinal strain every 6 mo 22 patients completed the study and were evaluable
Greater deterioration of strain in controls vs treatment group
Increased NT-proBNP in treatment group; not measured in controlsNCT01677286 Phase II, open-label, nonrandomized, prospective 25
Systemic amyloidosis (6 ATTRwt and 3 ATTRm)Doxycycline 100 mg every 12 h for 12 mo Drug safety
Response of affected organsWorsening of NT-proBNP and renal function
No improvement in other parameters studied
60% of patients had skin complications and 30% withdrew due to skin or gastrointestinal problemsNCT01171859 Phase II, open-label, nonrandomized, prospective 45
35 with heart involvement; 25 ATTRm; 5 ATTRm with TxH; 13 ATTRwt; and 2 domino liver transplant recipientsDoxycycline 100 mg every 12 h + TUDCA 250 mg every 8 h for 12 mo
Subsequent follow-up phase without treatment for 6 moDrug response defined as a < 2-point increase in NIS-LL and an increase < 30% or < 300 pg/mL NT-proBNP at 12 mo Cardiac response assessed in 25 patients
68% had cardiac response
Increased NT-proBNP and deterioration of strain during follow-up without treatment
High number of withdrawals due to adverse effects
14 withdrew in treatment phase and 5 discontinued in phase without treatmentEffect of doxycycline + UDCA on ATTR Phase II, open-label, nonrandomized, prospective 28
ATTR with cardiac involvement (27 ATTRm and 1 ATTRwt)Doxycycline 200 mg/d for 4 wk, then suspended 2 wk, then UDCA 750 mg/d for 12 mo
Subsequent follow-up phase without treatment for 6 moChanges in NT-proBNP and Kumamoto score Only 14% completed the study and 36% completed 12 mo
No changes in NT-proBNP at 6 mo and worsening at 12 mo
Stable LVH
Worsening of Kumamoto score at 12 moEGCG NCT01171859 Phase II, open-label, nonrandomized, prospective 25
ATTRwt600 mg, EGCG for 12 mo Changes in ECHO and cardiac MRI (n = 14) Decreased LV mass 6% by cardiac MRI (P = 0.03)
LVEF, myocardial thickness, and MAPSE by ECHO unchangedAntiSAP + CPHPC NCT03044353 Phase II, open-label, randomized 40
Cohort 1: cardiac ATTR amyloidosis
Cohort 2: primary amyloidosis after 6 mo of chemotherapyAnti-SAP + CPHPC monthly for 6 mo Reduced amyloid burden by cardiac MRI and ECHO Start in 2017 AntiSAP + CPHPC, antiserum amyloid P component + (R)-1-[6-[(R)-2-carboxy-pyrrolidin-1-yl]-6-oxo-hexanoyl]pyrrolidine-2-carboxylic acid; ATTRm, mutant transthyretin amyloidosis; ATTRwt, wild-type transthyretin amyloidosis; BNP, brain natriuretic peptide; ECHO, echocardiogram; EGCG, epigallocatechin-3 gallate; IV, intravenous; LV, left ventricle; LVEF, left ventricular ejection fraction; LVH, left ventricular hypertrophy; MAPSE, mitral annular plane systolic excursion; mNIS, Modified Neuropathy Impairment Score; MRI, magnetic resonance imaging; NIS, Neuropathy Impairment Score; NIS-LL, Neuropathy Impairment Score of the Lower Limbs; NT-proBNP, amino-terminal pro-brain natriuretic peptide; SC, subcutaneous; TTR, transthyretin; TUDCA, tauroursodeoxycholic acid; TxC, heart transplant; TxH, liver transplant; UDCA, ursodeoxycholic acid.
- •
The AOs are short strands of oligonucleotides that specifically bind to RNA, preventing translation and target protein synthesis.4 ISIS-TTRRX is a subcutaneous AO, with demonstrated dose-dependent reductions in TTR values of 75% to 90% in healthy volunteers.4 The phase III trial in patients with ATTRm and neurologic phenotype ended in March 2017 and its results are expected by the end of 2017. However, the US Food and Drug Administration postponed the initiation of a phase III trial in patients with ATTRwt and ATTRm with heart disease due to cases of severe thrombocytopenia in the neurologic study (Table 2). Since 50% of participants in the neurologic study had cardiac disease, the results of this cardiac substudy will determine if the phase III trial is resumed. On the other hand, there are preliminary data from an open-label phase II trial. In this study, 22 patients with ATTRwt and ATTRm with heart disease received a weekly injection of ISIS-TTRRX. According to report, the safety profile of the drug is very favorable and the intermediate data on cardiac disease progression by CMR, NT-proBNP, and 6-minute tests are positive.41
Dissociation of the TTR tetramer into subunits is a crucial step in ATTR fibril formation. Diflunisal and tafamidis are 2 TTR stabilizers with demonstrated efficacy in ATTRm polyneuropathy.
- •
Tafamidis is an orally administered small molecule that binds to TTR at T4 binding sites by stabilizing the protein and preventing its dissociation. Following publication of the results of a randomized double-blind trial in 125 patients with ATTRm and the Val30Met mutation in the initial stages of neurologic disease,42 the European Medicines Agency approved its use in 2011 as an orphan medicinal product to delay neurologic progression. Recent data demonstrate the effectiveness of the drug to achieve neurologic stability in at least 60% of participants after more than 4 years of follow-up. To date, it has limited use in ATTR and cardiologic disease. A phase II study in 21 patients with ATTRm and different mutations showed that NT-proBNP and echocardiographic parameters remained stable at 12 months.43 Data from a 5-year cohort study confirmed that the drug was well tolerated at a dose of 20 mg, although few patients with ATTRwt remained stable at 3.5 years.44 The ATTR-ACT trial is a 30-month phase III trial assessing the efficacy, safety, and tolerability of doses of 20 mg and 80 mg tafamidis vs placebo in 440 patients with ATTRm, ATTRwt, and HF. The primary endpoint includes hospital mortality and admission. Its results are expected in 2018.3,27
- •
Diflunisal is a nonsteroidal anti-inflammatory agent that stabilizes TTR molecules in vitro. It is not available in Spain, but can be medically requested from abroad for compassionate use. A phase III study of ATTRm in patients with predominantly neurologic involvement, more than half of whom had heart disease, found no significant differences in echocardiographic parameters over the study period (Table 2).45 Its potential for gastrointestinal adverse effects, kidney failure, water retention, and hypertension make it unsuited to patients with heart disease. Evidence on diflunisal in patients with ATTR is very limited. There is one study, but it was limited by its having a nonrandomized single-center design with little follow-up and few patients (n = 13). There were no admissions for decompensated HF, but there was significant worsening of renal function.46
- •
More recently, a Spanish group demonstrated that tolcapone (an oral catechol-O-methyltransferase inhibitor used in the treatment of Parkinson disease) has the ability to bind in vitro to the TTR of patients with ATTRwt and Val122Ile with higher affinity than other stabilizers.47
Amyloid deposits are very stable and it seems that the human organism has little ability to eliminate them. However, treatments that prevent new amyloid production, such as chemotherapy in AL, can gradually eliminate deposits at different organ-specific rates. Cardiac clearance is especially low and so far evidence of regression is scarce. Several molecules are currently under investigation to accelerate amyloid cardiac clearance in ATTR:
- •
Doxycycline (a commonly used antibiotic) disrupts the formation of amyloid fibrils. The synergistic effect of combined doxycycline and tauroursodeoxycholic bile acid (TUDCA), which is used in the treatment of liver disease, has been demonstrated to eliminate TTR deposits in animal models. A phase II study with 20 patients showed no cardiac or neurologic progression after 1 year of treatment with doxycycline/TUDCA, with an acceptable safety and tolerability profile.4 Other phase II studies have attempted to confirm these findings using combined doxycycline/TUDCA, doxycycline/ursodeoxycholic acid, or doxycycline alone.48–50 The preliminary results of one of these studies suggest a protective effect, with less worsening of cardiac function due to strain in the treatment group. Another of these studies obtained similar findings in 40 patients with ATTR: NT-proBNP, functional class, LVEF, and myocardial thickness parameters, among others, remained stable at 12 months (Table 2). Nevertheless, all these studies had a high dropout rate (35%-44%), mainly due to adverse effects, particularly sun hypersensitivity and gastrointestinal complaints (up to 30%).48–50
- •
The EGCG (epigallocatechin-3 gallate) is the most abundant catechin in green tea, and has been shown in vitro and in a murine model to inhibit amyloid formation and to eliminate existing deposits.4 The CMRI showed that daily administration of 600mg EGCG was associated with left ventricular mass stabilization in a group of 25 patients (Table 2).51
- •
The PRX004 is a monoclonal antibody that acts by binding to monomer-specific epitopes and misfolded TTR. It thus elicits elimination of deposits by activating phagocytosis.52 The basis of its mechanism of action is similar to that of an antibody used in AL. Phase II studies on this antibody are showing promising results. A phase I-II trial of this new antibody is set to begin in 2017.
- •
Regardless of the type of amyloid precursor protein, all deposits contain serum amyloid component P (SAP). Using this molecule as a target, anti-SAP antibodies have been shown to elicit a macrophage-mediated and complement-dependent reaction that caused major elimination of visceral amyloid deposits in a murine model. The bis-D-proline compound CPHPC can neutralize plasma SAP, and co-administration with anti-SAP IgG allows the antibody to reach SAP-containing deposits in tissue.53 A phase I study published in 2015 demonstrated elimination of hepatic deposits in 15 patients with systemic amyloidosis without cardiac involvement, with few adverse effects.53 A phase II study of patients with ATTR cardiac amyloidosis and AL is set to begin in 2017 (Table 2).
Transthyretin cardiac amyloidosis is diagnosed with increasing frequency. 99mTc-DPD scintigraphy and CMRI are examples of techniques that can be used for the simple and early identification of patients with ATTR.
Several ATTR-specific drugs are currently in the final phases of research. Therefore, we believe that ATTR cardiac amyloidosis will soon be considered a treatable entity rather than a fatal disease.
FUNDINGThis work was conducted with partial assistance from the Carlos III Health Institute and the Spanish Society of Cardiology (research grant 2016 to E. González-López). Assistance from the Carlos III Health Institute is funded by the European Regional Development Fund “Another Way to Make Europe”.
CONFLICTS OF INTERESTE. González-López has participated as a speaker in activities organized by Pfizer. P. Garcia-Pavia has received payments as a speaker in activities organized by Pfizer and as a consultant to Alnylam, Prothena, and Pfizer. E. González-López, A. López-Sainz, and P. Garcia-Pavia declare that Pfizer has funded research projects by their institution.