
Arrhythmogenic right ventricular cardiomyopathy (ARVC), first described in 1977 in patients undergoing surgical ablation for right ventricular outflow tract (RVOT) tachycardia,1 is a heart muscle disease characterized by cardiomyocyte loss and replacement with fibrous or fibro-fatty tissue that can result in arrhythmia, sudden cardiac death and heart failure. The disease is frequently inherited as an autosomal dominant trait caused by mutations in genes encoding for desmosomal2, 3, 4, 5, 6, 7, 8 and non-desmosomal9, 10, 11, 12, 13, 14 proteins. Analysis of disease expression in mutation carriers and their family members has shown that ARVC is a complex disease that makes clinical definition and diagnosis difficult. In 1994, diagnostic criteria were published by the Working Group on Myocardial and Pericardial Diseases of the European Society of Cardiology and the Scientific Council on Cardiomyopathies of the International Society and Federation of Cardiology.15 These were based on a scoring system that took into account right ventricular (RV) morphological and functional abnormalities, electrocardiographic (ECG) features, histopathological findings, ventricular arrhythmias and family history. These criteria were modified in 2010,16 to improve their sensitivity in early diagnosis and familial disease. This review will focus on the new ARVC criteria..
Historical perspectiveThe concept of a specific RV cardiomyopathy was first suggested in a report of six patients with sustained ventricular tachycardia (VT) and enlarged right ventricles published in 1977.1 In 1982, the term “arrhythmogenic right ventricular dysplasia” was first used in a case series of 24 patients with left bundle branch block (LBBB) pattern VT, RV wall motion abnormalities and replacement of the RV myocardium by adipose and fibrous tissue.17 However, arrhythmogenic right ventricular dysplasia (or as it was later renamed, ARVC) was not formally recognized as a distinct entity until 1994, following the publication of diagnostic criteria by the World Health Organization/International Society and Federation of Cardiology Task Force.15 These criteria were founded on the identification of structural abnormalities, fibrofatty replacement of the RV myocardium, characteristic ECG changes, arrhythmias of RV origin and evidence for familial disease..
The first gene mutations linked to the disease were identified in a recessive, syndromic variant of ARVC known as Naxos disease. The cause of this particularly malignant form of the condition was found to be a two-base pair deletion in the gene encoding plakoglobin,2 a major component of cell-to-cell junctions. This paved the way for the identification of disease-causing mutations in other genes encoding desmosomal proteins in the more common autosomal dominant forms of ARVC.3, 4, 5, 6, 7, 8 Latterly, very similar clinical and histological phenotypes have been identified in patients with mutations in non-desmosomal genes, including titin, desmin and lamin A/C.9, 10, 11, 12, 13, 14.
EpidemiologyThe prevalence of ARVC in the general population is difficult to estimate due to the challenging nature of the diagnosis. Studies in Europe report a prevalence of between 0.6 and 4.4 per 1000, but these may be biased by geographic variation in clinical and pathological expertise. ARVC is reported as a cause of sudden cardiac death in 11%-27% of individuals aged ≤35 years.18, 19, 20 The prevalence of sudden death in one Italian cohort was higher in athletes (22.4%) compared to non-athletes (8.2%).18.
2010 criteriaThe non-specific character of most clinical findings and the absence of a single diagnostic test, mean that the diagnosis of ARVC is always challenging. The extent of this challenge was first reflected in the 1994 International Task Force guideline that proposed standardised criteria for the diagnosis based upon the identification of structural, histological, ECG, arrhythmic and familial features, which were then subdivided into major and minor criteria according to their perceived specificity.15 The presence of two major criteria, one major plus two minor, or four minor criteria from different categories was considered diagnostic. Subsequent clinical experience with these guidelines suggested that, while the criteria are highly specific, they lack sensitivity for early disease. Many parameters were also subjective and not evidence based. For this reason, new criteria were proposed in 2010.16 The basic scheme is the same, but the diagnostic terminology for the revised criteria has been modified into the following:
• Definite diagnosis: two major, or one major and two minor or four minor criteria from different diagnostic categories..
• Borderline diagnosis: one major and one minor or three minor criteria from different diagnostic categories..
• Possible diagnosis: one major or two minor criteria from different diagnostic categories..
The following section considers the most relevant changes in the specific diagnostic domains of the new scheme..
ImagingSeveral structural and functional features of the right ventricle present important challenges for echocardiographic imaging (Figure 1); in particular, its position close to the anterior chest wall; its complex shape, orientation and geometry; and its thin wall. Compared to echocardiography, cardiovascular magnetic resonance (CMR) has several advantages which overcome some of these challenges. These include three dimensional visualisation of the heart and its relation to thoracic structures; the absence of geometrical assumptions in the quantification of RV and left ventricular (LV) volumes; excellent spatial resolution; and tissue characterisation. However, there are also limitations as the RV wall is very thin and partial volume effect may affect the image. In addition, differentiation between pericardial fat and fatty infiltration of the RV can be difficult. Finally, a lack of knowledge about the normal variability in RV morphology leads to high inter-observer and intra-observer variability in reporting..

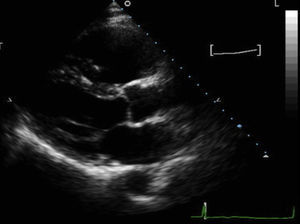
Figure 1. Parasternal long-axis view of a patient with arrhythmogenic right ventricular cardiomyopathy, showing severe right ventricular dilatation (right ventricular outflow tract 4.7cm); patient had severe right ventricular systolic dysfunction and akinetic right ventricular outflow tract wall.
The role of CMR in the diagnosis of ARVC has been recognised in the new Task Force criteria. While the 1994 criteria relied on qualitative assessment of the RV size and function (eg. “severe dilatation and reduction of RV ejection fraction” or “mild global RV dilatation and/or ejection fraction”), the new criteria introduce precise cut-offs for RV assessment. In addition, the presence of mild regional wall motion abnormalities (hypokinesia) has been excluded from the criteria, since it has very low specificity..
One of the major changes in the new imaging criteria is the recognition that LV involvement is part of the clinical spectrum of the disease. The 1994 criteria required little or no LV impairment to be present in order to preserve high specificity and to avoid inclusion of patients with dilated cardiomyopathy. Actually, several studies have shown that LV involvement is frequent with disease progression and may be a prognostic marker.21 Recently, forms with predominant LV involvement have been described,22 although here terminology becomes very confusing and somewhat arbitrary. In summary, the presence of LV dilatation and/or dysfunction is no longer an exclusion criteria for the diagnosis..
It should be noted that, while CMR can be used to assess fibrosis and fatty infiltration, its value in the thin RV wall is still debatable and tissue characterization is not part of the diagnostic criteria. On the other hand, the presence of late gadolinium enhancement and less consistently of myocardial fatty infiltration, particularly in the left ventricle, may help to identify affected individuals..
Tissue CharacterisationThe gold standard for the diagnosis of ARVC in the 1994 criteria was the demonstration of transmural fibro-fatty replacement of the RV myocardium on tissue obtained at necropsy, surgery or from endomyocardial biopsy.23 In this setting, endomyocardial biopsy has some intrinsic limitations, not least its failure to provide a transmural sample (important because the disease usually evolves from epicardium to endocardium). Moreover, endomyocardial biopsy samples are usually taken from the interventricular septum, which is less commonly involved than other regions of the RV. Nevertheless, histomorphometric parameters for the diagnosis of ARVC have been proposed: specifically, myocardial atrophy with residual myocytes<45% of the cross sectional area; fibrous tissue>40%; and fatty tissue>3%. These are reported to have a sensitivity of 67% and specificity of 92% for the diagnosis.23.
In a study on quantitative assessment of endomyocardial biopsy in ARVC using explanted hearts,24 there was considerable variability in the histological abnormalities between different sampling sites within the RV, with the antero-apical being the most informative region. Importantly, no useful diagnostic cut-off for fatty tissue was identified at the antero-apical and RVOT area. Parameters having a 95% specificity were: residual myocardium<59%, fibrosis>31% and fat>22%. The cut-off for fat was dependent on age and obesity and by excluding elderly and obese people groups a lower cut-off for fat was found (>9%). Based on these findings, new specific major and minor histological criteria have been proposed in the 2010 revision (Table 1)..
Table 1. 2010 Task Force Criteria for the Diagnosis of Arrhythmogenic Right Ventricular Cardiomyopathy
| I. Global and/or regional dysfunction and structural alterations | |
| Major | By 2-dimensional echocardiogram: regional RV akinesia, dyskinesia, or aneurysm and 1 of the following (end diastole):• PLAX RVOT≥32mm (corrected for body size [PLAX/BSA]≥19 mm/m2)• PSAX RVOT≥ 36mm (corrected for body size [PSAX/BSA]≥21 mm/m2)• O fractional area change≤33% |
| By MRI: regional RV akinesia or dyskinesia or dyssynchronous RV contraction and 1 of the following:• Ratio of RV end-diastolic volume to BSA≥110 mL/m2 (male) or ≥100 mL/m2 (female)• O RV ejection fraction≤40% | |
| By RV angiography: regional RV akinesia, dyskinesia, or aneurysm | |
| Minor | By 2-dimensional echocardiogram: regional RV akinesia or dyskinesia and 1 of the following (end diastole):• PLAX RVOT≥29 to <32mm (corrected for body size [PLAX/BSA]≥16 to <19 mm/m2)• PSAX RVOT≥32 to <36mm (corrected for body size [PSAX/BSA]≥18 to <21 mm/m2)• O fractional area change >33% to ≤40% |
| By MRI: regional RV akinesia or dyskinesia or dyssynchronous RV contraction and 1 of the following:• Ratio of RV end-diastolic volume to BSA≥100 to < 110 mL/m2 (male) or ≥90 to <100 mL/m2 (female)• Or RV ejection fraction >40% to ≤45% | |
| II. Tissue characterisation of the wall | |
| Major | Residual myocytes<60% by morphometric analysis (or <50% if estimated), with fibrous replacement of the RV free wall myocardium in ≥1 sample, with or without fatty replacement of tissue on endomyocardial biopsy |
| Minor | Residual myocytes 60% to 75% by morphometric analysis (or 50% to 65% if estimated), with fibrous replacement of the RV free wall myocardium in ≥1 sample, with or without fatty replacement of tissue on endomyocardial biopsy |
| III. Repolarisation abnormalities | |
| Major | Inverted T waves in right precordial leads (V1, V2, and V3) or beyond in individuals >14 years of age (in the absence of complete RBBB QRS≥120 ms) |
| Minor | Inverted T waves in leads V1 and V2 in individuals>14 years of age (in the absence of complete RBBB) or in V4, V5, or V6Inverted T waves in leads V1, V2, V3, and V4 in individuals>14 years of age in the presence of complete RBBB |
| IV. Depolarisation/conduction abnormalities | |
| Major | Epsilon wave (reproducible low-amplitude signals between end of QRS complex to onset of the T wave) in the right precordial leads (V1 to V3) |
| Minor | Late potentials by SAECG in ≥1 of 3 parameters in the absence of a QRS duration of ≥110ms on the standard ECG: filtered QRS duration (fQRS)≥114 ms; duration of terminal QRS<40μV (low-amplitude signal duration)≥38 ms; root-mean-square voltage of terminal 40 ms≤20μVTerminal activation duration of QRS≥55ms measured from the nadir of the S wave to the end of the QRS, including R’, in V1, V2, or V3, in the absence of complete RBBB |
| V. Arrhythmias | |
| Major | Nonsustained or sustained ventricular tachycardia of left bundle-branch morphology with superior axis (negative or indeterminate QRS in leads II, III, and aVF and positive in lead aVL) |
| Minor | Nonsustained or sustained ventricular tachycardia of RVOT configuration, LBBB morphology with inferior axis (positive QRS in leads II, III, and aVF and negative in lead aVL) or of unknown axis>500 ventricular extrasystoles per 24h (Holter) |
| VI. Family history | |
| Major | ARVC/D confirmed in a first-degree relative who meets current Task Force criteriaARVC/D confirmed pathologically at autopsy or surgery in a first-degree relativeIdentification of a pathogenic mutation * categorized as associated or probably associated with ARVC/D in the patient under evaluation |
| Minor | History of ARVC/D in a first-degree relative in whom it is not possible or practical to determine whether the family member meets current Task Force criteriaPremature sudden death (<35 years of age) due to suspected ARVC/D in a first-degree relativeARVC/D confirmed pathologically or by current Task Force criteria in second-degree relative |
ARVC/D, arrhythmogenic right ventricular cardiomyopathy/dysplasia; aVF, augmented voltage unipolar left foot lead; aVL, augmented voltage unipolar left arm lead; BSA, body surface area; ECG, electrocardiogram; LBBB, left bundle branch block; MRI, magnetic resonance imaging; PLAX, parasternal long-axis view; PSAX, parasternal short-axis view; RBBB, right bundle branch block; RV, right ventricular; RVOT, right ventricular outflow tract; SAECG, signal-averaged electrocardiogram.
* A pathogenic mutation is a DNA alteration associated with ARVC/D that alters or is expected to alter the encoded protein, is unobserved or rare in a large non-ARVC/D control population, and either alters or is predicted to alter the structure or function of the protein or has demonstrated linkage to the disease phenotype in a conclusive pedigree.Modified from Marcus et al. 16 with permission of the editor.
Recently, a new immunohistochemical tool for the diagnosis of ARVC, that determines localization of plakoglobin on endomyocardial biopsy, has been proposed.25 The test, however, is not specific for ARVC, as it is also positive in patients with sarcoidosis and giant cell myocarditis.26.
Repolarisation AbnormalitiesThe ECG plays a key role in the diagnosis of ARVC (Figure 2) and ECG abnormalities have been emphasised in the revised criteria. One of the key features of ARVC is the presence of T-wave inversion in right precordial leads, in absence of complete right bundle branch block (RBBB). The prevalence of T-wave inversion in right precordial leads in healthy individuals beyond 14 years of age has been found only in 4% of women and 1% of men.27 Therefore this criterion has now been considered specific enough to be a major abnormality, while in the 1994 criteria it was minor. In addition, the presence of T-wave inversion in V1-V2 (in individuals beyond 14 years of age and in the absence of complete RBBB) has been introduced as a minor abnormality. The recognition that LV involvement can occur or even be the main characteristic in ARVC is also reflected in the repolarisation criteria, which included the presence of T-wave inversion in lateral leads, V4, V5 o V6 (typically seen in cases of LV involvement) as minor criterion..

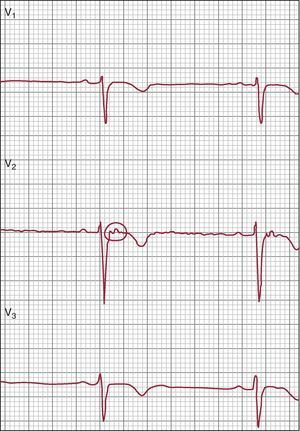
Figure 2. Electrocardiogram from a patient with arrhythmogenic right ventricular cardiomyopathy, showing sinus rhythm, poor R-wave progression and T-wave inversion in right precordial leads.
A study by Jain et al.28 systematically assessed the value of ECG features in ARVC, including patients with incomplete and complete RBBB. In patients with ARVC and complete RBBB, the only two parameters that differed compared to a control population were the presence of T-wave inversion up to V4 (59% vs 12%, respectively) and r’/s ratio in V1 < 1 (88% vs 14%, respectively). Based on these observations, the presence of T-wave inversion in V1-V4 in individuals beyond 14 years of age with complete RBBB is considered a minor criterion..
Depolarisation/Conduction AbnormalitiesEpsilon waves (Figure 3) are electrical potentials of small amplitude that occur at the end of the QRS complex and at the beginning of the ST segment detected in the right precordial leads. Their identification can be facilitated using a high pass filter of 40Hz, increasing the gain to 20mV/mm and the recording speed to 50mm/s. A modification of the limb lead position (right arm lead on the manubrium sternum, left arm lead on the xyphoid process and left leg lead on a rib between the usual V4-V5 position) may improve sensitivity.30 Epsilon waves are thought to represent areas of delayed activation in the right ventricle as a consequence of fibrous and/or fibro-fatty replacement of RV myocardium and are considered a major criterion..

Figure 3. Epsilon wave in V2 (circle). Reproduced from Quarta et al. 29 with the permission of the editor.
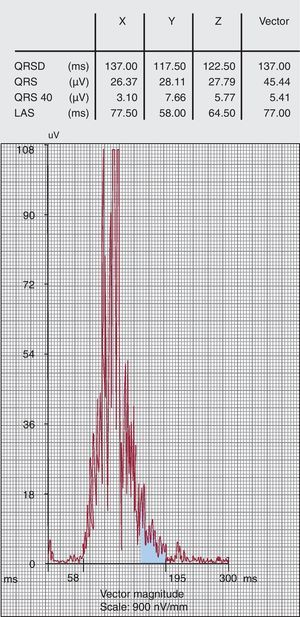
The presence of late potentials detected by signal-averaged ECG (Figure 4) suggests areas of slow ventricular conduction which may represent an anatomical substrate for re-entrant ventricular arrhythmias. Signal-averaged ECG is performed using a 40-250Hz filter in order to assure a noise level<0.3μV. Three parameters are routinely analysed: filtered QRS duration, duration of low amplitude signal (<40μV) of the terminal QRS (LAS); and root mean square amplitude of the last 40 ms of the QRS signal (QRS 40). In the original criteria, late potentials were considered present if two of these parameters were abnormal: specifically, filtered QRS≥114 ms, LAS≥38 ms and QRS40≤20μV. However, these values were not evidence based. The sensitivity and specificity of the signal-averaged ECG has been compared in 69 ARVC patients and 103 normal controls16: each parameter had a sensitivity of 58%-60% and a specificity of 94%-96%, while two parameters lead to similar sensitivity (66%) and specificity (95%); using any of the parameters gave a sensitivity of 74% and specificity of 92%. Based on these observations, one or more parameters (in the absence of a QRS duration≥110ms) is considered a minor criterion in the new diagnostic algorithm..

Figure 4. Signal-averaged electrocardiogram positive for late potentials. LAS, duration of low amplitude signal (<40μV) of the terminal QRS; QRS 40, root mean square amplitude of the last 40 ms of the QRS signal; QRSD, filtered QRS duration.
Nasir et al.31 proposed a new criterion, which also represents delayed conduction in the RV myocardium: terminal activation duration of the QRS≥55 ms measured from the nadir of the S wave to the end of the QRS in V1, V2 o V3, in the absence of complete RBBB. In their cohort, this new minor criterion was present in 95% of patients with ARVC (including 20% where it was the only ECG abnormality detected) and only 7% of patients with RVOT tachycardia and 2% of normal controls..
ArrhythmiasVentricular arrhythmias are a common finding in patients with ARVC and often represent the initial presentation of the disease. Sustained and non-sustained VT and ventricular ectopics typically have a LBBB morphology reflecting their RV origin (Figure 5). When LV involvement is present, ventricular arrhythmias with a RBBB morphology may be seen..

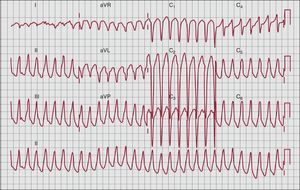
Figure 5. Left-bundle branch block ventricular tachycardia with inferior axis (right ventricular outflow tract origin).
The RVOT is also the site of origin for idiopathic RVOT VT, a benign entity occurring in patients with structurally normal hearts. The new ARVC criteria take this into account in order to increase specificity of ventricular arrhythmia. In the case of VT with LBBB morphology and an inferior axis (positive QRS in leads II, III and aVF and negative QRS in aVL), the VT is a minor criteria, since it can be found also in idiopathic RVOT tachycardia. In contrast, VT with LBBB configuration and a superior axis (negative or indeterminate QRS in leads II, III and aVF and positive QRS in aVL) is more specific for ARVC and it is now considered a major criterion. If the axis of VT is unknown, then the arrhythmia is a minor criterion..
The presence of more than 500 ventricular ectopics in 24 h tape (reduced from 1000/24h in 1994 criteria) is a minor criterion..
Family History and GeneticsThe clinical diagnosis of ARVC in an individual changes the genetic risk to their first degree relatives from 1:1000-1:5000 in the general population to 1:2. The new ARVC criteria acknowledge this by making unequivocal disease in a first degree relative a major criterion (while in the 1994 criteria it was a minor criterion). If the clinical diagnosis of ARVC is in a second degree relative or if there is history of ARVC in a first degree relative, but it is not possible to determine if the family member meets Task Force criteria, then it represents a minor criterion. By the same principle, a history of ARVC confirmed pathologically at autopsy or surgery is a major criterion if the family member is a first degree relative, otherwise is a minor criterion. As in the 1994 criteria, a premature young (<35 years of age) sudden cardiac death due to suspected ARVC is a minor criterion..
Five genes encoding important desmosomal proteins are thought to account for disease in up to 70% of patients.32 A number of non-desmosomal gene mutations have been associated with ARVC. These include: the ryanodine receptor 2 gene (RYR2)11; mutations in the 5’ and 3’ untranslated regions of the transforming growth factor beta 3 (TGFβ3) gene12; the transmembrane protein 43 (TMEM43)9; desmin10; and, most recently, titin (TTN)13 and lamin A/C genes.14 The new ARVC criteria include the presence of a pathogenic mutation associated or probably associated with ARVC in an individual with clinical suspicion of the disease. The definition of pathogenicity of the mutation is strict and relies on the demonstration that the variant is not present or rare in healthy control populations, alters or is predicted to alter the function and/or structure of the encoded protein and/or has been shown to cosegregate with the disease. Strict adherence to these criteria is essential, as variants in desmosomal genes seem relatively common in normal individuals and their expression is highly variable. Indeed, many patients have more than one pathogenic variant, suggesting that genetic load is particularly important in this disease..
ConclusionsOf all the cardiomyopathies, ARVC is the most difficult to describe in simple and clinically useful terms. The new ARVC criteria represent a step forward in the attempt to identify individuals affected by the disease, but even now, most diagnoses will reflect a variable degree of probability that depends very much on the clinical context. Most importantly, the revised criteria should be considered a dynamic process since knowledge about ARVC, the genetic basis and pathogenic mechanisms is constantly evolving..
Conflicts of interestNone declared..
.
Corresponding author: The Heart Hospital, 16-18 Westmoreland Street, London W1G 8PH, United Kingdom. perry.elliott@ucl.ac.uk