

Our objective was to approximate the prevalence of mutations in candidate genes for familial hypercholesterolemia (FH) in a middle-aged Spanish population and to establish the predictive value of criteria for clinical suspicion in the detection of causative mutations.
MethodsUnrelated individuals aged ≥ 18 years from the Aragon Workers’ Health Study (AWHS) with high low-density lipoprotein cholesterol (LDL-C) and clinical suspicion of FH (participants with LDL-C concentrations above the 95th percentile, participants with premature cardiovascular disease and/or participants with high LDL-C [130 mg/dL] under statin therapy), assuming that any participant with FH exhibits at leats 1 trait, were selected and the LDLR, APOB, PCSK9, APOE, STAP1 and LDLRAP1 genes were sequenced by next generation sequencing technology.
ResultsOf 5400 individuals from the AWHS, 4514 had complete data on lipid levels and lipid-lowering drugs, 255 participants (5.65%) met the criteria for suspicion of FH, 24 of them (9.41%) were diagnosed with hyperlipoproteinemia(a), and 16 (6.27% of those sequenced) were found to carry causative mutations in candidate genes: 12 participants carried 11 different pathogenic LDLR alleles and 4 participants carried 1 pathogenic mutation in PCSK9. LDL-C concentrations> 220 mg/dL and LDL-C> 130 mg/dL despite statin therapy showed the strongest association with the presence of mutations (P=.011).
ConclusionsOur results show that the prevalence of FH in Spain is 1:282 and suggest that the combination of high untreated LDL-C and high levels of LDL-C despite statin therapy are the best predictors of a positive FH genetic test.
Keywords
Familial hypercholesterolemia (FH) is a genetic disorder characterized by very high plasma total cholesterol concentrations, due to increased low-density lipoprotein cholesterol (LDL-C), with a high risk of premature coronary heart disease (CHD).1 Traditionally, FH has been described as a monogenic disease, with autosomal codominant transmission and an estimated prevalence of around 1:500 in the general population.2 However, there are some discrepancies in the prevalence of FH. Recent studies have revealed that clinically defined FH is probably more common than previously reported, with a prevalence of 1:217 in the Copenhagen General Population study.3 Early diagnosis is crucial in FH because lipid-lowering treatment has been demonstrated to drastically reduce CHD,4 especially if treatment begins early in life.5 However, FH is underdiagnosed and undertreated in the general population.6
The clinical diagnosis of FH relies on high plasma LDL-C, family history of hypercholesterolemia, personal and family history of premature CHD, and signs of cholesterol deposition such as tendon xanthomas and premature arcus cornealis. These variables are often clinically scored by applying the Make Early Diagnosis to Prevent Early Death (MEDPED) criteria, the Dutch Lipid Clinic Network (DLCN) MEDPED modification, or the Simon Broome Register Group (SBRG) criteria.7 These clinical criteria have been demonstrated to be strongly associated with genetic diagnosis.8,9 However, these criteria include information that is not always available or requires some expertise, and they are mostly used in specialized units.6,10 For this reason and to improve diagnosis, the European Atherosclerosis Society Consensus Panel recommends the criteria for suspicion prior to clinical diagnosis based on the following: a) for adults, plasma total cholesterol above 310 mg/dL or the 95th percentile by age and sex for country; b) for children, plasma total cholesterol level above 230mg/dL or the 95th percentile by age and sex for country; c) premature CHD (< 55 years in men and <60 years in women); d) tendon xanthomas; e) sudden premature CHD death in a family member (< 55 years in men and <60 years in women)6. However, the clinical value of these criteria for suspicion has not been validated.
Molecular genetic diagnosis is the gold-standard in a monogenic disease and is strongly recommended in FH.11 FH is caused by mutations in several genes: LDLR, coding for the low-density lipoproteins (LDL) receptor; APOB, coding for apolipoprotein B,12 and PCSK9, coding for the enzyme proprotein convertase subtilisin/kexin type 9.13 One new locus causing FH has been identified: the p.(Leu167del) mutation in APOE.14 In addition, several mutations in the signal transducing adaptor family member STAP1 have been associated with FH,15 although the role of this gene has recently been questioned.16,17 Furthermore, plasma levels of lipoprotein(a) (Lp(a)) is an inheritable trait associated with increased CHD risk, and it has been reported that hyperlipoproteinemia(a) (hyperLpa(a)) is a common cause of autosomal dominant hypercholesterolemia.18 Knowledge of the causative mutation confirms diagnosis, confers prognosis, facilitates family screening, and improves patient adherence to treatment.19 The presence of pathogenic mutations in FH causative genes in patients with a clinical diagnosis of FH confers a substantially increased risk for CHD,20 and therefore genetic tests allow identification of persons with the highest CHD risk among people with hypercholesterolemia. However, this genetic testing also has some difficulties, including the genetic complexity of the disease with several genes involved, thousands of different mutations, some of them of uncertain pathogenic significance, and the poor availability of genetic analysis in some health systems.
The objective of the present study was to approximate the prevalence of mutations in candidate genes in a middle-aged Spanish population based on criteria for clinical suspicion and to analyze their predictive value for the presence of causative mutations.
METHODSParticipantsIndividuals were selected from the Aragon Workers’ Health Study (AWHS). The AWHS is a longitudinal cohort study of cardiovascular risk factors and subclinical atherosclerosis performed among 5400 workers at the Opel Spain factory in Aragon, Spain, who have been followed up since 2009.21 Here, we studied unrelated individuals ≥ 18 years old from the AWHS, with annual or biannual determinations since 2009. The following criteria were used to select those with clinical suspicion of FH: a) participants with untreated LDL-C concentrations> 95th percentile of the Spanish population stratified by age and sex22 in at least 2 measurements; b) participants with premature CHD (< 55 years in men and <60 years in women) at baseline; c) participants under statin therapy and LDL-C> 130 mg/dL in at least 2 measurements; d) participants with untreated LDL-C concentrations> 95th percentile of the Spanish population in just 1 measurement but who also had LDL-C> 130mg/dL under statin therapy. The exclusion criteria were the presence of secondary causes of hypercholesterolemia at baseline or during follow-up, which include severe obesity (body mass index [BMI]> 35 kg/m2), poorly controlled type 2 diabetes (HbA1c> 8%), renal disease with glomerular filtration rate <30mL/min and/or macroalbuminuria, liver disease (alanine transaminase> 3 times the upper normal limit), hypothyroidism (thyroid stimulating hormone> 6 mIU/L), pregnancy, autoimmune diseases, and treatment with protease inhibitors. We defined hyperLp(a) participants as those participants with suspicion of FH who had Lp(a)> 50 mg/dL, but normal LDL-C concentrations adjusted by Lp(a) (< 95th percentile of the Spanish population)22.
All participants gave written informed consent, and the study was approved by the Aragón Ethics Committee for Clinical Research (CEICA).
Assessment of cardiovascular risk factors, personal and family history of cardiovascular disease, intake of drugs affecting lipid metabolism, and anthropometric measurements were performed in all participants at baseline and annually or biannually during the follow-up.
Lipid analysisLipid and lipoprotein analyses were performed on EDTA plasma samples collected after an overnight fast of at least 10hours annually or biannually since 2009. Measurements up to 2015 were included. Total cholesterol and triglyceride levels were determined by standard enzyme methods. High-density lipoprotein cholesterol (HDL-C) was measured directly by an enzyme reaction using cholesterol oxidase (UniCel DxC 800; Beckman Coulter, Inc, USA). LDL-C was calculated by Friedewald's formula. Lipid values were adjusted according to statin therapy in those participants taking lipid-lowering drugs.23 None of these participants received PCSK9 inhibitors. Lp(a) was determined by IMMAGE kinetic nephelometry (Beckman Coulter, Inc, USA). LDL-C adjusted by Lp(a) was obtained by subtracting the concentrations of Lp(a) divided by 3 to the LDL-C concentration calculated by the Friedewald formula.24,25
Genetic analysisWhole blood genomic DNA was isolated using standard methods. LDLR (NM_000527.4), PCSK9 (NM_174936.3), APOE (NM_000041.3), STAP1 (NM_0121108.3), and LDLRAP1 (NM_015627.2) genes and exons 26 and 29 of APOB (NM_000384.2), which encodes the LDLR binding site, were sequenced by next generation sequencing with the SeqProLipo Platform (Progenika Biopharma Grifols, Spain). This platform includes point mutations, large rearrangements, and copy number variations.
To evaluate the pathogenicity of identified variants we used SIFT,26 PolyPhen-2,27 Mutation Taster,28 and PredictSNP.29 The effect of variants in potential splicing sites was predicted with FruitFly.30 To compare the frequency of identified variants in the study participants with that of the general population, we compiled the allele frequencies of the identified variants from the 1000 Genomes Project31 and ExAc Browser Data.32 Rare variants were defined as genetic variants with a frequency <1% in the general population. A rare variant was defined as a causative variant when it had been previously associated with FH or possibly pathogenic when the bioinformatic analysis prediction classified it as pathogenic.
Statistical analysisAnalyses were performed using statistical computing software R version 3.5.0.33 The distribution of the variables was analyzed with the Shapiro test. Quantitative variables with a normal distribution are expressed as mean± standard deviation and were analyzed with the ANOVA test. Variables with a skewed distribution are expressed as median and interquartile range and were analyzed with the Kruskal-Wallis test. Qualitative variables are expressed as percentages and were analyzed with the chi-square test. The level of significance was set at P <.05. Logistic binary regression was used to determine the best European Atherosclerosis Society criteria diagnosis with the presence of a mutation in candidate genes as the dependent variable.
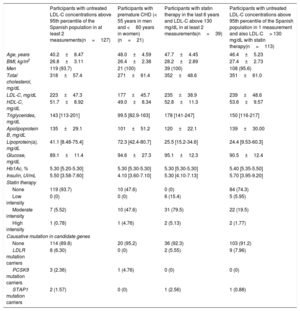
RESULTSStudy participants meeting FH clinical criteriaOf the 5400 individuals from the AWHS, 4514 participants had the information required by the inclusion and exclusion criteria. A total of 886 individuals were excluded because they had incomplete recorded lipid data, less than 2 complete lipid panel and/or incomplete information about lipid-lowering drug use. Of the 4514 participants, 255 (5.65%) met the following inclusion criteria (not mutually exclusive): a) 127 participants had untreated LDL-C concentrations> 95th percentile of the Spanish population in at least 2 measurements; b) 21 participants had premature CHD at baseline (< 55 years in men and <60 years in women); c) 39 participants had LDL-C> 130mg/dL in at least 2 measurements while taking statin therapy and; d) 113 participants had untreated LDL-C concentrations> 95th percentile of the Spanish population in 1 measurement and also had LDL-C> 130mg/dL on a least another occasion with statin therapy (figure 1).

Participants with high levels of LDL-C despite statin therapy and those with CHD at baseline were older and had higher BMI than the rest of the participants. Participants with statin therapy and high levels of LDL-C despite drug treatment, together with participants with high LDL-C concentrations in 1 measurement and also high levels of LDL-C with statin therapy, had higher concentrations of total cholesterol, LDL-C and triglycerides than the remaining participants. Participants with premature CHD had higher levels of Lp(a). The percentage of participants with high levels of LDL-C in at least 2 measurements who did not take any lipid-lowering therapy was 93.7% (table 1).
Baseline biochemical and clinical characteristics, and number of mutation carriers according to selection criteria.
| Participants with untreated LDL-C concentrations above 95th percentile of the Spanish population in at least 2 measurements(n=127) | Participants with premature CHD (< 55 years in men and <60 years in women)(n=21) | Participants with statin therapy in the last 6 years and LDL-C above 130 mg/dL in at least 2 measurements(n=39) | Participants with untreated LDL-C concentrations above 95th percentile of the Spanish population in 1 measurement and also LDL-C> 130 mg/dL with statin therapy(n=113) | |
|---|---|---|---|---|
| Age, years | 40.2±8.47 | 48.0±4.59 | 47.7±4.45 | 46.4±5.23 |
| BMI, kg/m2 | 26.8±3.11 | 26.4±2.38 | 28.2±2.89 | 27.4±2.73 |
| Men | 119 (93.7) | 21 (100) | 39 (100) | 108 (95.6) |
| Total cholesterol, mg/dL | 318±57.4 | 271±61.4 | 352±48.6 | 351±61.0 |
| LDL-C, mg/dL | 223±47.3 | 177±45.7 | 235±38.9 | 239±48.6 |
| HDL-C, mg/dL | 51.7±8.92 | 49.0±8.34 | 52.8±11.3 | 53.6±9.57 |
| Triglycerides, mg/dL | 143 [113-201] | 99.5 [82.9-163] | 178 [141-247] | 150 [116-217] |
| Apolipoprotein B, mg/dL | 135±29.1 | 101±51.2 | 120±22.1 | 139±30.00 |
| Lipoprotein(a), mg/dL | 41.1 [8.48-75.4] | 72.3 [42.4-80.7] | 25.5 [15.2-34.6] | 24.4 [9.53-60.3] |
| Glucose, mg/dL | 89.1±11.4 | 94.6±27.3 | 95.1±12.3 | 90.5±12.4 |
| Hb1Ac, % | 5.30 [5.20-5.30] | 5.30 [5.30-5.30] | 5.30 [5.30-5.30] | 5.40 [5.35-5.50] |
| Insulin, UI/mL | 5.50 [3.58-7.60] | 4.10 [3.60-7.10] | 5.30 [4.10-7.13] | 5.70 [3.95-9.20] |
| Statin therapy | ||||
| None | 119 (93.7) | 10 (47.6) | 0 (0) | 84 (74.3) |
| Low intensity | 0 (0) | 0 (0) | 6 (15.4) | 5 (5.95) |
| Moderate intensity | 7 (5.52) | 10 (47.6) | 31 (79.5) | 22 (19.5) |
| High intensity | 1 (0.78) | 1 (4.76) | 2 (5.13) | 2 (1.77) |
| Causative mutation in candidate genes | ||||
| None | 114 (89.8) | 20 (95.2) | 36 (92.3) | 103 (91.2) |
| LDLR mutation carriers | 8 (6.30) | 0 (0) | 2 (5.55) | 9 (7.96) |
| PCSK9 mutation carriers | 3 (2.36) | 1 (4.76) | 0 (0) | 0 (0) |
| STAP1 mutation carriers | 2 (1.57) | 0 (0) | 1 (2.56) | 1 (0.88) |
BMI, body mass index; CHD, coronary heart disease; HbA1c, glycosylated hemoglobin; HDL-C, High-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol. Total, LDL-C and HDL-C and triglyceride levels have been adjusted according to statin therapy.23 Selected participants may belong to more than one group.
Data are expressed as No. (%), mean±standard deviation or median [interquartile range].
Sequencing analysis reported 5280 variants in candidate genes among the 255 sequenced participants (figure 1): a) 1792 variants in LDLR, 11 of which have been described as pathogenic and previously associated with FH; b) 249 variants in APOB, although none of them has been described as pathogenic or previously associated with FH; c) 1697 variants in PCSK9, only 1 of which has been described as pathogenic and previously associated with FH; d) 72 variants in APOE, 2 of which have been previously associated with type III hyperlipoproteinemia; e) 869 variants in STAP1, 3 of which have been described as possibly pathogenic by bioinformatic analysis; f) 601 variants in LDLRAP1 gene, 2 of which have been classified as possibly pathogenic by bioinformatic analysis. However, neither of these 2 possibly pathogenic variants in LDLRAP1 were present in homozygosity, a condition that has been described as necessary to cause hypercholesterolemia.
The percentage of mutations in candidate genes was similar among groups; however, participants with high levels of LDL-C in at least 2 measurements showed the highest percentage of causative mutations, exceeding 8% of cases (table 1). A total of 12 patients were carriers of 11 distinct pathogenic alleles in heterozygosity in the LDLR: 1 of them with a causative variant located in the promoter region (c.-135C>G); 7 of them had a causative variant, located in coding regions, producing an amino acid change (c.530C>T, c.826T>G, c.862G>A, c.1247G>A, c.1529C>T, c.1775G>A, and c.1816G>A); 1 of them with a causative variant, located in an intronic region, producing an alternative splicing change (c.1586+5G>A); 1 allele had 2 causative variants located in cis: (c.274C>G) and (c.313+1G>C) and 1 of them had a large rearrangement (c.941-?_1845+?del). All of them have been described as pathogenic by bioinformatic analysis and have been previously reported as a cause of FH.34–40 Only 1 patient carried a rare variant in exon 26 of the APOB gene, (c.10621A>G). However, this variant has not been previously associated as a cause of FH, has been classified as benign by bioinformatic analysis, and seems not to affect the binding region structure. A total of 5 patients carried 2 rare variants in the PSCK9 gene (c.60_65dup GCTGCT and c.743G>A), and only the first one has been previously associated with FH. Four of these patients were carriers of the c.60_65dupGCTGCT in-frame indel, which has been described as uncertain or likely pathogenic by Clin Var41 and Garcia et al.42 A total of 5 patients carried 3 rare variants in the APOE gene, none of them previously described as a cause of FH. One of them has not been previously associated with any hyperlipidemia and has been classified as unknown by bioinformatic analysis (c.335C>A). The other 2 variants, (c.460C>A, c.487C>T), were present in 3 and 1 participants, respectively. Both variants have been previously associated with type III hyperlipoproteinemia.43,44 A total of 4 patients carried 3 unknown rare variants in the STAP1 gene; 2 of them carried 1 variant located at the 5’ region (c.-60A>G) and the other 2 were carriers of 2 different variants, located in coding regions, which produce amino acid substitution (c.619G>A and c.803T>C). None of these variants have been previously associated with FH. Two patients carried 2 unknown rare variants in heterozygous state in the LDLRAP1 gene, 1 of them producing an amino acid change (c.605C>G) and the other could affect splicing (c.748-7C>G). However, mutations in LDLRAP1 only produce hypercholesterolemia when they are found in homozygosity (table 2 and ).
Causative variants in candidate genes identified in patients selected in this study
| Gene | SNV | Nucleotide | Amino acid change | Number of carriers | Bioinformatic analysis | Clin Var | Frequency ExAc32 | Frequency 1000 G31 | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| SIFT26 | POLYPHEN-227 | Mutation Taster28 | PredictSNP29 | ||||||||
| LDLR | rs879254375 | c.-135C>G | NA | 1 participant | NA | NA | NA | NA | Pathogenic | - | - |
| LDLR | rs774467219rs112029328 | c.[274C>G;313+1G>C] | p.[Gln92Glu;NA] | 1 participant | Tolerated (0.15) | Possiblydamaging (0.736) | Deleterious(0.510) | Neutral(0.252) | Pathogenic | 0.0009715 | 0.001 |
| LDLR | rs121908026 | c.530C>T | p.(Ser177Leu) | 1 participant | Deleterious(0.01) | Probablydamaging (0.999) | Deleterious(0.896) | Deleterious (0.000090) | Pathogenic | 8.958e-06 | - |
| LDLR | rs879254692 | c.826T>G | p.(Cys276Gly) | 1 participant | Deleterious(0) | Probablydamaging(0.969) | Deleterious(0.856) | Deleterious (0.000005) | Pathogenic | - | - |
| LDLR | rs368657165 | c.862G>A | p.(Glu288Lys) | 1 participant | Deleterious(0.05) | Probably damaging (0.918) | Deleterious (0.714) | Deleterious (0.000018) | Pathogenic | 0 | - |
| LDLR | - | c.941-?_1845+?del | Deletion from exon 7 to exon 12 | 1 participant | Pathogenic | ||||||
| LDLR | rs773658037 | c.1247G>A | p.(Arg416Gln) | 1 participant | Deleterious(0.01) | Probably damaging (0.957) | Deleterious(0.806) | Deleterious (0.000045) | Pathogenic | 1.793e-05 | 0 |
| LDLR | rs755154048 | c.1529C>T | p.(Thr510Met) | 1 participant | Deleterious (0.02) | Possibly damaging (0.791) | Deleterious(0.679) | Deleterious (0.000013) | Pathogenic/Likely pathogenic | 8.952e-06 | - |
| LDLR | rs781362878 | c.1586+5G>A | NA | 1 participant | NA | NA | NA | NA | Uncertain/Pathogenic | 1.796e-05 | |
| LDLR | rs137929307 | c.1775G>A | p.(Gly592Glu) | 2 participants | Deleterious(0.01) | Probably damaging(0.925) | Deleterious(0.779) | Deleterious (0.000015) | Pathogenic | 8.951e-05 | - |
| LDLR | rs72658865 | c.1816G>A | p.(Ala606Thr) | 1 participant | Deleterious(0.02) | Possibly damaging(0.5) | Deleterious (0.550) | Deleterious (0.000034) | Likely benign/ Uncertain | 8.952e-06 | - |
| PCSK9 | rs371488778 | c.60_65dupGCTGCT | p.(Leu22_Leu23dup) | 4 participants | NA | NA | NA | Deleterious (0.000002) | Uncertain/ Pathogenic | 0.002144 | - |
NA, not applicable.
In 24 (9.41%) participants with criteria for suspicion of FH due to LDL-C> 95th percentile, when the cholesterol transported in Lp(a) was analyzed, their LDL-C were no longer> 95th percentile. None of these hyperLp(a) participants was a carrier of a pathogenic mutation in FH candidate genes.
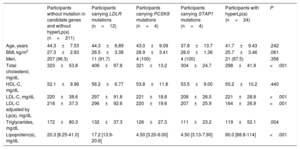
Characteristics according to genetic analysisTable 3 shows the clinical characteristics and lipid profile according to the genetic classification including hyperLp(a) participants. Participants carrying LDLR mutations had the highest levels of total and LDL-C. In addition, participants carrying PCSK9 mutations had significantly higher levels of total and LDL-C than those carrying STAP1 mutations and participants classified with hyperLp(a) (P <.001 and P <.001, respectively). Moreover, participants carrying STAP1 mutations had the lowest levels of total and LDL-C, and had lower levels of total and LDL-C than participants without mutation in candidate genes.
Clinical characteristics and lipid profile according to the primary cause of hypercholesterolemia
| Participants without mutation in candidate genes and without hyperLp(a) (n=211) | Participants carrying LDLR mutations (n=12) | Participants carrying PCSK9 mutations (n=4) | Participants carrying STAP1 mutations (n=4) | Participants with hyperLp(a) (n=24) | P | |
|---|---|---|---|---|---|---|
| Age, years | 44.3±7.53 | 44.3±6.89 | 43.0±9.09 | 37.8±13.7 | 41.7±9.43 | .242 |
| BMI, kg/m2 | 27.3±2.83 | 26.5±3.38 | 28.9±3.41 | 26.0±1.36 | 25.7±3.46 | .061 |
| Men, | 207 (96.3) | 11 (91.7) | 4 (100) | 4 (100) | 21 (87.5) | .356 |
| Total cholesterol, mg/dL | 323±53.8 | 406±97.8 | 321±13.2 | 304±24.7 | 298±41.9 | <.001 |
| HDL-C, mg/dL | 52.1±9.96 | 56.2±6.77 | 53.8±11.8 | 53.5±9.00 | 55.2±10.2 | .440 |
| LDL-C, mg/dL | 220±38.6 | 297±91.8 | 221±18.8 | 208±26.0 | 221±28.8 | <.001 |
| LDL-C adjusted by Lp(a), mg/dL | 218±37.3 | 296±92.6 | 220±19.6 | 207±25.9 | 164±26.9 | <.001 |
| Triglycerides, mg/dL | 172±80.3 | 132±37.3 | 126±27.3 | 111±23.2 | 119±52.1 | .004 |
| Lipoprotein(a), mg/dL | 20.3 [8.25-41.0] | 17.2 [13.9- 20.6] | 4.50 [3.20-6.00] | 4.50 [3.13-7.90] | 80.0 [68.8-114] | <.001 |
BMI, body mass index; HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; Lp(a), lipoprotein(a).
The P value was calculated by ANOVA test or Kruskal-Wallis and chi-square, as appropriate.
Data are expressed as No. (%), mean±standard deviation or median [interquartile range].
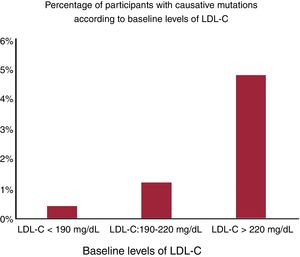
Mutations in the LDLR gene were responsible for 75% of some mutations in candidate genes, vs 25% of those in participants who carried some mutation in the PSCK9 gene. Figure 2 shows the percentage of mutations according to groups by baseline LDL-C levels. The higher the percentage, the greater the increase in LDL-C: 0.40% of participants with baseline LDL-C <190mg/dL, 1.20% of participants with baseline LDL-C from 190-220mg/dL, and 4.80% of participants with baseline LDL-C> 220mg/dL had a mutation in candidate genes. Considering those patients with a mutation in candidate FH genes plus those with hyperLp(a), almost 16% of the group with criteria for suspicion had monogenic disease.

Logistic binary regression was used to study the association of clinical data with 2 response variables: mutation in any gene and mutation in the LDLR gene. The analysis showed that baseline LDL-C, LDL-C adjusted by Lp(a) and total cholesterol levels were significantly associated with the presence (yes/no) of any mutation in candidate genes (OR, 1.023; P <.001; OR, 1.024; P <.001 and OR, 1.0.17; P <.001 respectively, table 4A-C). Nevertheless, no criterion showed a significant association with the presence of any mutation in candidate genes. The presence of mutation in the LDLR (yes/no) was significantly associated with baseline total and LDL-C (OR, 1.009; P=.010 and OR, 1.0.16; P=.0488, respectively) and the criteria for suspicion of FH: untreated LDL-C concentrations above the 95th percentile in 1 measurement and also LDL-C> 130mg/dL while taking statins (P=.011, table 4D).
Logistic binary regression with presence of mutation in candidate genes as dependent variable
| A | OR | 95%CI | P | R2 Nagelkerke |
|---|---|---|---|---|
| Baseline age, years | 0.962 | 0.879-1.065 | .243 | 0.192 |
| Baseline BMI, kg/m2 | 1.018 | 0.838-1.234 | .852 | |
| Baseline LDL-C, mg/dL | 1.023 | 1.012-1.036 | <.001 |
| B | OR | 95%CI | P | R2 Nagelkerke |
|---|---|---|---|---|
| Baseline age, years | 0.958 | 0.873-1.061 | .379 | 0.213 |
| Baseline BMI, kg/m2 | 1.026 | 0.839-1.25 | .797 | |
| Baseline LDL adjusted by Lp(a), mg/dL | 1.024 | 1.013-1.037 | <.001 |
| C | OR | 95%CI | P | R2 Nagelkerke |
|---|---|---|---|---|
| Baseline age, years | 0.961 | 0.880-1.059 | .389 | 0.157 |
| Baseline BMI, kg/m2 | 0.995 | 0.821-1.204 | .959 | |
| Baseline total cholesterol, mg/dL | 1.017 | 1.008-1.025 | <.001 |
| D | OR | 95%CI | P | R2 Nagelkerke |
|---|---|---|---|---|
| CRITERION 1a | 2.332 | 0.822-7.591 | .127 | 0.099 |
| CRITERION 2b | 0.735 | 0.039-4.153 | .775 | |
| CRITERION 3c | 0.324 | 0.026-7.607 | .389 | |
| CRITERION 4 | 4.01 | 1.163-18.40 | .011 |
95%CI, 95% confidence interval; BMI, body mass index; LDL, low-density lipoprotein; LDL-C, low-density lipoprotein cholesterol; Lp(a), lipoprotein(a); OR, odds ratio.
Participants with untreated LDL-C concentrations above 95th percentile of the Spanish population in one measurement and also LDL-C> 130mg/dL with statin therapy.
Table 4A-C show the logistic binary regression using the presence of any mutation (yes/no) in any candidate gene. Table 4D shows the logistic binary regression using the presence of any mutation (yes/no) in the LDLR gene.
Table 5 shows the number of participants with a mutation based on the criteria they met, positive and negative predictive values and sensitivity and specificity regarding the sample selected due to the presence of at least 1 criterion. The first criterion had the highest positive predictive value (10.2%) to detect the presence of functional mutations in candidate genes (LDLR, PCSK9 and STAP1 genes), especially to detect mutations in the LDLR and PCSK9 genes (6.3% and 2.4%). Nevertheless, the fourth criterion showed the highest positive predictive value to detect mutations in the LDLR gene (8.0%), showing in this selected sample higher specificity than the first criterion (0.510 and 0.572, respectively).
Number of participants carrying a mutation based on the criteria they meet
| Participants who meet criteria | PPV | NPV* | Sensibility* | Specificity* | ||||
|---|---|---|---|---|---|---|---|---|
| CRITERION 1 | Yes (n=127) | No (n=128) | ||||||
| Mutation | Yes | No | Yes | No | ||||
| Participants with mutation | 13 | 114 | 7 | 121 | 0.102 | 0.945 | 0.650 | 0.514 |
| Participants with LDLR mutation | 8 | 119 | 4 | 124 | 0.063 | 0.969 | 0.667 | 0.510 |
| Participants with PCSK9 mutation | 3 | 124 | 1 | 127 | 0.024 | 0.992 | 0.750 | 0.506 |
| Participants with STAP1 mutation | 2 | 125 | 2 | 126 | 0.016 | 0.984 | 0.500 | 0.498 |
| CRITERION 2 | Yes (n=21) | No (n=234) | PPV | NPV* | Sensibility* | Specificity* | ||
|---|---|---|---|---|---|---|---|---|
| Mutation | Yes | No | Yes | No | ||||
| Participants with mutation | 1 | 20 | 19 | 215 | 0.048 | 0.919 | 0.050 | 0.915 |
| Participants with LDLR mutation | 0 | 21 | 12 | 222 | 0.000 | 0.949 | 0.000 | 0.914 |
| Participants with PCSK9 mutation | 1 | 20 | 3 | 231 | 0.048 | 0.987 | 0.750 | 0.920 |
| Participants with STAP1 mutation | 0 | 21 | 4 | 230 | 0.000 | 0.983 | 0.000 | 0.916 |
| CRITERION 3 | Yes (n=39) | No (n=216) | PPV | NPV* | Sensibility* | Specificity* | ||
|---|---|---|---|---|---|---|---|---|
| Mutation | Yes | No | Yes | No | ||||
| Participants with mutation | 3 | 36 | 17 | 199 | 0.077 | 0.921 | 0.150 | 0.847 |
| Participants with LDLR mutation | 2 | 37 | 10 | 206 | 0.051 | 0.954 | 0.166 | 0.848 |
| Participants with PCSK9 mutation | 0 | 39 | 4 | 212 | 0.000 | 0.981 | 0.000 | 0.845 |
| Participants with STAP1 mutation | 1 | 38 | 3 | 213 | 0.026 | 0.986 | 0.250 | 0.849 |
| CRITERION 4 | Yes (n=113) | No (n=142) | PPV | NPV* | Sensibility* | Specificity* | ||
|---|---|---|---|---|---|---|---|---|
| Mutation | Yes | No | Yes | No | ||||
| Participants with mutation | 10 | 103 | 10 | 132 | 0.088 | 0.930 | 0.500 | 0.562 |
| Participants with LDLR mutation | 9 | 104 | 3 | 139 | 0.080 | 0.979 | 0.750 | 0.572 |
| Participants with PCSK9 mutation | 0 | 113 | 4 | 138 | 0.000 | 0.972 | 0.000 | 0.550 |
| Participants with STAP1 mutation | 1 | 112 | 3 | 139 | 0.009 | 0.979 | 0.250 | 0.553 |
NPV, negative predictive value; PPV, positive predictive value.
This study analyses the frequency of pathogenic mutations in candidate genes for monogenic FH in a population with clinical suspicion of FH. Three major conclusions can be drawn from our results. First, high LDL-C is the main factor associated with a positive genetic diagnosis; second, a high LDL-C alone is not specific enough to be used for FH identification, requiring genetic analysis, and third, Lp(a) concentration should be included in the diagnostic algorithm for FH. Our study analyses more than 4500 individuals from the AWHS, a healthy middle-aged population, showing that approximately 5% (255 participants) fulfilled the criteria for diagnostic suspicion of FH, but only 16 participants (0.4%) had a pathogenic mutation in the LDLR or PCSK9 genes.
FH identification is an important issue because FH mutation carriers have a substantially increased risk for CHD,20 and therefore genetic study allows identification of the highest risk within hypercholesterolemic participants.5 The analysis of candidate genes together with Lp(a) quantification allows identification of almost 16% of the causes of these severe hypercholesterolemia groups. Our data are in agreement with previously published reports on the prevalence of hyperLp(a) as a cause of primary hypercholesterolemia18,45 and reinforces the idea that genetic evaluation of patient with suspected FH should include candidate genes and Lp(a) concentration quantification.9
The concept of FH is evolving and, possibly with the current clinical criteria, the disease is a genetic severe hypercholesterolemia syndrome, which is sometimes monogenic, and sometimes polygenic or with a complex background.46 This article refers to heterozygous FH, which is the most common form within FH with a definite clinical diagnosis and the form most closely associated with CVD, and which therefore requires an earlier presumptive diagnosis.
Traditionally, the prevalence of FH has been estimated at 1:500.2 However, subsequent studies have revealed that clinically defined FH is probably more common than previously reported, with a prevalence of 1:217 in the Copenhagen General Population study.3 In our study, we found that 255 participants, from a total of 4514 individuals studied, met the criteria for suspicion of FH, which indicates that 1:18 met the criteria for suspicion of FH. Of 255 participants, 20 participants had a rare variant in the LDLR, PCSK9, or STAP1 genes, which would suggest a prevalence of FH of 1:226. Of 20 participants with rare variants in candidate genes, 12 of them had a mutation in LDLR, 4 of them were carriers of a mutation in PCSK9, and 4 of them had a variant in STAP1.
According to recent studies,16,17,47 the role of STAP1 has not been clearly associated with FH phenotype and, for that reason, we recalculated the prevalence of FH using only carriers of mutations in LDLR and PCSK9 genes. In this way, the prevalence would decrease from 1:226 to 1:282. In the present study, we have demonstrated that LDLR FH carriers have the extreme FH phenotype, as reported previously.48,49 Therefore, if we take into account only LDLR carriers, the prevalence of FH decreases from 1:226 to 1:376.
Sequencing analysis of LDLR, APOB, PCSK9, APOE, STAP1 and LDLRAP1 reported 5280 variants, but only 16 of them were possibly pathogenic or pathogenic according to in silico analysis: a) eleven pathogenic variants in the LDLR gene previously associated with FH,34–40b) 1 pathogenic variant in the PCSK9 gene (c.60_65dupGCTGCT), which has a frequency less than 0.5% in the general population and has been classified as pathogenic by bioinformatic analysis. However, further studies are needed to investigate the functionality of this variant; c) 1 rare variant in APOB (c.10621A>G). This missense variant, which produces an amino acid change, p.(Ile3515Val), in the mature protein, is located in the beta 2 domain of apolipoprotein B and computational prediction tools and conservation analyses suggest that this variant would not impact protein function. In addition, computational splicing tools suggest that this variant would not impact the RNA splicing50; d) 3 rare variants in the STAP1 gene; 1 of them located in the 5’UTR (c.-60A>G), and 2 of them located in coding regions (c.619G>A and c.803T>C), which produce amino acid change, p.(Ile268Thr) and p.(Asp207Asn), respectively. All these variants have been classified as pathogenic by bioinformatic analysis; however, none of them has previously been associated with FH. Further research is needed to investigate the role of STAP1 in FH phenotype, as previous studies have reported participants carrying STAP1 mutations with normal levels of total and LDL-C and incomplete association with FH; 16,17,51e) 2 rare variants in LDLRAP1 gene, 1 of them located in intronic region (c.748-7C>G), which could produce an alternative splicing, and another one located in coding region (c.605C>G), producing an amino acid change, p.(Ser202Cys). This missense variant has been classified as pathogenic by bioinformatic analysis. However, the individual carried this variant in heterozygosity, which would not explain the FH phenotype; f) 3 missense rare variants in APOE gene, 2 of them (c.460C>A and c.487C>T) described as pathogenic by Clin Var52 and associated with type III hyperlipoproteinemia,43,44 but not with FH.
Our results showed that LDL-C levels were significantly associated with the presence of a causative mutation in candidate genes. While in our sample from a working population CHD was small and it had the smallest positive predict value for mutations, participants with untreated LDL-C concentrations above> 220mg/dL and those with LDL-C above 130 mg/dL despite statin therapy showed a significant association with the presence of a pathogenic mutation. Among them, the probability of finding a causative mutation in candidate gene was 4 times higher than that in patients meeting the remaining criteria for suspicion of FH. In the future, the progressive decline in the costs of DNA analysis will probably facilitate universal screening programs in the population. However, because of their current costs, candidates must be selected for genetic testing. Our results suggest that not only high levels of untreated LDL-C are a good marker of FH in the general population, but the combination of these levels with high levels of LDL-C despite statin therapy could be a better predictor of FH. Furthermore, participants with high levels of untreated LDL-C and high levels of LDL-C with statin therapy showed the highest percentage of mutations in LDLR (7.96%), and these participants also had a more extreme FH phenotype.
The percentage of mutations in candidate genes varies widely, from 55.6% when a history of family monogenic pattern of hypercholesterolemia information is available,8 to less than 2% in the case of exclusive use of LDL levels> 190 mg/dL.20 Our study fully agrees with the concept that an isolated high LDL-C without information of family history of severe hypercholesterolemia allows identification of a small percentage of FH.
LimitationsOur study has some limitations. First, AWHS is a cohort with a high prevalence of men, obesity, hypertension, and hypercholesterolemia, which may not be representative of the general population. However, because it is a relatively young population there, will be no bias in the prevalence of FH mutations. Second, we report some mutations, c.743G>A in the PCSK9 gene and c.10621A>G in APOB, with uncertain significance. However, both are described as neutral or benign by bioinformatic analysis and the ClinVar database, so there are not enough data to classify them as pathogenic variants. Sequencing analysis of the APOB gene included only exon 26 and exon 29, while sequencing the entire APOB gene could report other variants as a cause of FH phenotype. Nevertheless, hypercholesterolemia due to APOB defects would require that the protein would have defective binding to the LDL receptor, since mutations causing only lower expression or improper folding of ApoB would produce hypocholesterolemia. Therefore, the variants causing FH are expected to be located in the coding region of the ApoB binding domain (in exon 26 and 29). In fact, most APOB mutations described as a cause of FH have been identified in exon 26.49 Finally, possibly pathogenic mutations in the PCSK9 gene (c.60_65dupGCTGCT) and in the STAP1 gene (c.-60A>G, c.619G>A and c.803T>C) need further studies to gain greater insight into their role in the FH phenotype.
CONCLUSIONSThe present study analyses the positive predictive value of clinical diagnostic criteria for FH. Our study included 4514 individuals from the AWHS, a healthy middle-aged population. A total of 255 individuals met the diagnostic criteria for clinical suspicion of FH, of whom 16 (6.27%) had mutations in the LDLR and PCSK9 genes, which corresponds to a prevalence of FH of 1:282 in this population. Furthermore, 24 participants (9.41%) were diagnosed with hyperlipoproteinemia(a), which let us to identify almost 16% of the etiology of these severe hypercholesterolemia groups. Untreated LDL-C concentrations and high levels of LDL-C despite statin therapy showed a significant association with the presence of mutations in candidate genes. These results suggest that the combination of high untreated LDL-C with high levels of LDL-C despite statin therapy could be a first step, but not the only one, for FH screening in the population.
FUNDINGThis work was supported by grants from Gobierno de Aragón, B14-7R, Spain, and the Spanish Ministry of Economy and Competitiveness PI15/01983, PI18/01777 and CIBERCV. These projects are co-financed by Instituto de Salud Carlos III and the European Regional Development Fund (ERDF) of the European Union “A way to make Europe”. CIBERCV is a project of Instituto de Salud Carlos III.
CONFLICTS OF INTERESTThe authors declare no conflicts of interest.
- –
FH is a genetic disorder characterized by high plasma total and LDL cholesterol concentrations and high risk of CVD.
- –
Clinical criteria have been demonstrated to be highly associated with genetic diagnosis.
- –
However, 20% to 40% of patients with the FH phenotype do not have a mutation in candidate genes.
- –
The European Atherosclerosis Society has recommended new diagnostic criteria for suspicion of FH, which have not yet been validated.
- –
The prevalence of FH in Spain is 1:282.
- –
Only 6.27% of participants with suspected FH had functional mutations in the LDLR and PCSK9 genes.
- –
A total of 9.41% of participants with suspected FH were diagnosed with hyperlipoproteinemia(a).
- –
The combination of high untreated LDL-C with high levels of LDL-C despite statin therapy is the best predictor for a positive finding of FH mutation.
Supplementary data associated with this article can be found in the online version available at https://doi.org/10.1016/j.rec.2020.06.003