Heart failure due to antineoplastic therapy remains a major cause of morbidity and mortality in oncological patients. These patients often have no prior manifestation of disease. There is therefore a need for accurate identification of individuals at risk of such events before the appearance of clinical manifestations. The present article aims to provide an overview of cardiac imaging as well as new “-omics” technologies, especially with regard to genomics and proteomics as promising tools for the early detection and prediction of cardiotoxicity and individual responses to antineoplastic drugs.
Keywords
Cardiotoxicity induced by antineoplastic drugs is becoming an important health problem for oncological patients treated with traditional agents (anthracyclines and cyclophosphamide), as well as new agents (monoclonal antibodies and tyrosine kinase inhibitors).1 The majority of these patients have no prior disease manifestations.2 In addition, the conventional indexes and biomarkers of cardiotoxicity often show evident changes only after cardiac damage has occurred.3 There is consequently a need for accurate identification of individuals at risk of heart disease before the appearance of clinical manifestations. The identification of new genes and signaling pathways by the “-omics” approach in clinical practice may be useful to identify early cardiac damage and new therapeutic targets.4,5 In this article, we provide an overview of cardiac imaging as well as new “-omics” technologies, especially with regard to genomics and proteomics as promising tools for the early detection and prediction of cardiotoxicity and individual responses to antineoplastic drugs.
ANTICANCER DRUG-INDUCED CARDIOTOXICITY: DEFINING THE PROBLEMCardiotoxicity is defined as the appearance of cardiac muscle dysfunction due to exposure to antineoplastic therapy, which may progress to heart failure (HF). Subclinical cardiotoxicity, or preclinical cardiotoxicity, refers to the early stage of this cardiomyopathy, when the disease is not clinically evident. Since the current diagnosis of cardiotoxicity is still based on the onset of HF symptoms or a decrease in left ventricular ejection fraction (LVEF), and because of interobserver variability in LVEF measurement, the incidence of cardiotoxicity may vary, depending on the type of antiblastic therapy, as well as on the type of detection system used to formulate the diagnosis.6 The use of high-sensitivity cardiac troponin-I and new echocardiographic parameters, such as strain/strain rate, as well as the use of new biomarkers capable of identifying patients at risk of developing heart disease, can help to make an early diagnosis of the disease. The antineoplastic drugs most commonly used to treat cancer are anthracyclines, antimetabolites, human epidermal growth factor receptor 2 (HER2) receptor inhibitors, and tyrosine kinase inhibitors. Anthracyclines (ie, doxorubicin and epirubicin), mainly used for the treatment of breast cancer and hematologic neoplasms, induce irreversible cardiotoxicity in a dose-dependent manner and through generation of free radicals, DNA damage, and cell death of cardiomyocytes and cardiac progenitor cells. In a retrospective analysis by Von Hoff et al.,7 the percentage of patients who developed left ventricular dysfunction (detected by echocardiographic estimation of LVEF) at a cumulative doxorubicin dose of 400mg/m2 was 3%, increasing to 7% at 550mg/m2, and to 18% at 700mg/m2. A retrospective analysis of 3 prospective trials8 evaluating LVEF by multiple-gated acquisition (MUGA) nuclear scans, found that 5% of patients developed left ventricular dysfunction at a cumulative dose of 400mg/m2, 26% of patients did so at 550mg/m2, and 48% of patients did so at 700mg/m2. Inhibitors of the HER2 receptor (ie, trastuzumab), are monoclonal antibodies mainly used for the treatment of breast and gastric cancers, which induce reversible cardiotoxicity through HER2 blockade in cardiomyocytes and cardiac progenitor cells, regardless of the dose used. In patients treated with trastuzumab, symptomatic HF can occur in between 1.7 and 20.1%.9 Tyrosine kinase inhibitors (ie, sunitinib) are mainly used for the treatment of renal cell carcinoma, are capable of inducing symptomatic HF through mitochondrial injury and cardiomyocyte apoptosis, with an incidence ranging from 4.1% to 33.8%.10
ROLE OF THE REDOX (REDUCTION/OXIDATION) BALANCE IN CARDIOTOXICITYThe cardiotoxic mechanisms of several antiblastic agents, including anthracyclines, tyrosin kinase inhibitors and antimetabolites, can involve oxidative stress due to insufficient inactivation of reactive oxygen species (ROS), as well as increased generation of ROS by xanthine oxidases, NAD(P)H oxidases (NOXs) and mitochondrial complexes I and III.11–15 In particular, electrons leaked from mitochondrial complexes I and III can represent the main source of superoxide anions (O2−).16,17 Antiblastic agents may activate myocardial NOX2, normally quiescent, which in turn produces O2−, whereas NOX4, which is constitutively active, generates hydrogen peroxide (H2O2). Oxidative stress is further amplified by the conversion of ROS to the more toxic hydroxyl radical (OH–) by several processes within and outside mitochondria, including reaction of nitric oxide (NO) with subsequent formation of reactive nitrogen species (RNS).13 NO is generated by endothelial (eNOS or NOS3) and neuronal (nNOS or NOS1) nitric oxide synthases, which are constitutively expressed in cardiomyocytes, as well as by inducible NOS2 (iNOS), which is stimulated by proinflammatory mediators or ischemic preconditioning.18–20 NO can also be produced by other reactions, collectively called “non-NOS” processes. These include reactions catalyzed by “non-NOS enzymes”, such as cytochrome c, hemoglobin, and xanthine oxidoreductase, and reactions due to “nonenzymatic” processes under acidic conditions, such as the reduction of nitrite to NO. Nitrite and NO can produce different biological actions by direct or indirect posttranslational nitration (3-nitrotyrosine formation) or the nitrosation/nitrosylation of specific targets, such as metals and cysteine thiol residues.16–20 NO and ROS lead to the formation of RNS, including peroxynitrite (ONOO−). High levels of ROS and RSN may damage cardiovascular cells or may impact the cellular signaling pathways in the cardiovascular system. In particular, ROS can lead to membrane lipid peroxidation and DNA damage, with subsequent membrane damage and apoptotic cell death. Stimulated ROS/RNS and NOX-related damage contribute to both the initiation and progression of many solid and hematopoietic cancers,21 whereas antiblastic drugs, and in particular anthracyclines, may induce cardiotoxicity via altered ROS/RNS production and/or an endogenous antioxidant system disturbance within the cardiovascular system.22,23 The specific redox alterations induced by anthracyclines can be traced back to the generation of unstable metabolites (such as doxorubicin-semiquinone), which in turn can react with O2, producing H2O2 and O2−. In addition, anthracyclines chelate the free intracellular iron, forming iron-doxorubicin complexes. These can react with O2, leading to the generation of ROS. Furthermore, anthracyclines can directly interfere with the main iron-transporting and -binding proteins,24 resulting in mitochondrial iron accumulation and further generation of ROS. Finally, ROS interact with cardiolipin, a mitochondrial membrane phospholipid involved in apoptotic pathways, leading to the release of mitochondrial apoptogenic factors, such as cytochrome c. Since NO can block cardiolipin oxidation by inhibiting the peroxidase activity of cytochrome c on the cardiolipin complex, site-specific and appropriate amounts of NO may counteract the toxic effects of anthracyclines.25,26 Oxidative stress may also play a role in cardiotoxicity derived from tyrosin kinase inhibitors: the direct infusion of sunitinib in different experimental preparations provoked a dose-dependent cardiodepressant effect, accompanied by decreased levels of intracellular Ca2+, with a concomitant rise in ROS generation.27 In addition, 5-fluorouracil such as capecitabine and gemcitabine, can induce oxidative stress in cardiomyocytes and endothelial NOS dysregulation, endothelin 1 upregulation, and the activation of protein kinase C. These effects may lead to endothelium-dependent and -independent vasoconstriction, and subsequently to coronary spasms.28–30 Although oxidative stress is essential for anthracycline-induced cardiotoxicity, clinical studies have shown that blocking redox reactions by antioxidant agents is not cardioprotective.2,6 The failure of these agents can be attributed to the multifactorial etiology of cardiotoxicity, which is not exclusively caused by oxidative stress. Another reason for the failure is the physiological role of ROS/RSN, which is altered by antioxidant agents. Therefore, it is important to develop intelligent and sophisticated therapies that can alter the redox system at key points, without disturbing the physiological role of oxidative stress. Nevertheless, any new drugs require experiments in appropriate in vitro and animal models and comparative studies with other already approved cardioprotective drugs (eg, dexrazoxane), as well as randomized trials in the context of antiblastic cardiotoxicity.31,32
DETECTION AND MONITORING CARDIOTOXICITY BY CARDIAC IMAGINGCardiac imaging of patients who undergo chemotherapy is an essential step in the early diagnosis of cardiotoxicity once the damage, although initial, has occurred.33,34 Combinations of cardiac imaging modalities that integrate the strengths of each modality and at the same time eliminate the weaknesses of an individual modality could offer improved diagnosis and therapeutic monitoring of cardiotoxicity.34 However, during chemotherapy, the use of different cardiac imaging techniques, such as echocardiography, MUGA, and cardiac magnetic resonance to evaluate LV volumes and function in the same patient, is not recommended due to the significant difference in results across the techniques. Therefore, choosing a single tool for serial monitoring of LV function during chemotherapy is preferred. Because of the requirement of radiation exposure and low capability to provide comprehensive information about right ventricular function and the presence of valvular or pericardial disease, MUGA has a low impact as an imaging technique for the diagnosis of cardiotoxicity. Because of its safety, wide availability, repeatability, and low cost, 2-dimensional (2D) echocardiography is considered the first-line method to assess ventricular function alterations and to stratify the risk of HF and manage treatments.35 The criteria to diagnose cardiomyopathy due to anticancer therapy aim to identify the global or regional decrease of systolic function, with a decline in LVEF of ≥ 5% to < 55%, or a decrease in LVEF of > 10 percentage points.36 New parameters, such as strain/strain rate, have been developed for an earlier detection of LV dysfunction. In addition, the echocardiographic evaluation of diastolic function by prolongation of the isovolumic relaxation time and tissue Doppler imaging, may be useful to detect cardiac damage by cancer therapy.37 Since chemotherapy can lead to an increased risk of endocarditis, especially in cancer patients who are immunocompromised and more prone to nonbacterial thrombotic lesions, due to malignancy-associated thrombophilia,38 systematic assessment of valvular vegetations and severity of valve regurgitations is required. Moreover, irradiation of the heart can cause the a complication known as radiation-induced heart disease, which includes valvular stenosis or regurgitation.39 Transthoracic 2D-echocardiography should be the first-line examination in patients with suspicion of valve damage due to cancer therapy, while transesophageal echocardiography is the gold standard for a more detailed diagnosis.36 While LV dysfunction and HF may be common in cancer patients after antineoplastic therapy, clinical evidence of right HF is extremely rare, although some drugs, like anthracycline, cyclophosphamide, and 5-fluorouracil, can induce impairment of right ventricular systolic and diastolic function.40 Therefore, all patients receiving antineoplastic therapy should undergo echocardiographic evaluation of the right ventricle with the following measurements: basal diameter and area, tricuspid annular plane systolic excursion, peak tricuspid annulus systolic velocity by tissue Doppler imaging, and fractional area change.41 Transthoracic echocardiography is also the method of choice for the evaluation of the pericardium, which may be damaged by cancer therapy. Echocardiographic findings in these patients can be entirely normal or show clear evidence of pericardial effusion. Echocardiography allows the diagnosis of pericardial effusion and cardiac tamponade, as well as guidance of pericardiocentesis.42 Cardiac magnetic resonance should be considered when echocardiographic information is unsatisfactory or when tissue characterization is needed.6,43
DETECTION AND PREDICTION CARDIOTOXICITY BY THE “-OMICS” APPROACHThe genomic and proteomic data available to date are limited to cardiac toxicity induced by conventional antineoplastic agents such as antracyclines and antimitotic agents. Since the importance of ROS/RNS production as early mediators of chemotherapy-related cardiotoxicity, biomarkers with redox significance—known as biomarkers of oxidative/nitrosative cardiotoxicity—can be identified by the “-omics” approach. This approach may offer novel tools for the identification of early markers of cardiotoxicity and the development of innovative cardioprotective agents.4,5 A decrease in nicotinamide adenine dinucleotide phosphate (NAD(P)H):quinone oxidoreductase 1 activity and an increase in ROS production by NAD(P)H oxidases have been considered early biomarkers of antracycline-induced cardiotoxicity.44 A significant variation in ROS has been observed together with a change in the enzymatic activity of glutathione peroxidases, which was correlated with an early variation in longitudinal systolic function in patients after the administration of epirubicin.45 Genetic variants can allow identification of the individual variability of the response to antineoplastic drugs, which may be essential for personalized medicine and to decrease the adverse effects of chemotherapy.46,47 Significant single nucleotide polymorphisms (SNPs) associated with a higher risk of developing anthracycline-induced cardiotoxicity included those within NAD(P)H oxidase as well as doxorubicin efflux transporter genes.48,49 In a case-control study by Wojnowski et al.,48 109 patients with non-Hodgkin lymphoma treated with anthracycline and 363 controls were screened for SNPs related to 82 genes involved in the generation of ROS. The authors identified 5 significant SNP in NAD(P)H oxidase associated with a higher risk of developing cardiotoxicity. In particular, chronic cardiotoxicity was associated with a variant of the NAD(P)H oxidase p40phox subunit (NCF4 gene, SNP rs1883112, -212A>G), while acute cardiotoxicity was associated with a SNP of the NAD(P)H oxidase p22phox subunit (CYBA gene, SNP rs4673 c.242C>T).48 These mutations lead to missense variation His72Tyr, associated with a dysfunction of the NAD(P)H oxidase. The involvement of SNP related to NAD(P)H oxidase in the development of cardiotoxicity was also demonstrated by Cascales et al.49 who observed a strong association between rs1883112 SNP NCF4 and cardiac interstitial fibrosis. Finally, the role of NCF4 rs1883112 SNP as an independent predictor of cardiotoxicity was confirmed by a subsequent study, conducted in patients with diffuse large B-cell lymphoma, treated with R-CHOP21 (rituximab with cyclophosphamide, DXR, vincristine, and prednisone).50 Other SNPs, equally important as contributors to anthracycline-induced cardiotoxicity, either through the generation of free radicals or toxic metabolites, have been identified among genes coding for proteins involved in the P450 oxidoreductase gene.51–53 In a case-control study by Lubieniecka et al.,52 286 patients with acute myeloid leukemia treated with daunorubicin, were screened for SNPs (rs2868177, rs13240755 and rs4732513) in the P450 oxidoreductase (POR) gene. These SNPs were significantly associated with a decrease in LVEF after daunorubicin administration.52 Wasielewski et al.54 also conducted genetic screening in 6 families with dilated cardiomyopathy (DCM), showing the presence of 3 mutations (c.1633G>A, p.Asp545Asn; c.2863G>A, p.Asp955Asn and c.4125T>A, p.Tyr1375X) in the MYH7 sarcomeric gene in 2 patients with anthracycline-induced DCM and a positive family history of DCM. Visscher et al.46 screened 2 cohorts of 156 and 188 anthracycline-treated children for 2977 SNPs in 220 key drug biotransformation genes. The authors identified a highly significant association of SNP rs7853758 (c.1381C>T) within the SLC28A3 gene that conferred significant protection against cardiotoxicity induced by anthracycline.
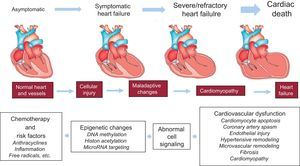
Fewer data are available on the epigenetics of antineoplastic drug-induced cardiotoxicity.55–61 Epigenetic modifications such as cytosine and histone modifications are heritable genomic features that do not change the DNA sequence. They are involved in the regulation of the expression of protein-encoding genes and noncoding RNAs (miRNAs), which are noncoding RNAs involved in the posttranscription regulation of gene expression.59 In addition, a specific group of miRNAs (defined as epi-miRNAs) can directly target effectors of the epigenetic machinery, such as DNA methyltransferases (DNMTs), histone deacetylase (HDACs) or polycomb genes, and indirectly affect the expression of genes, whose expression is controlled by epigenetic factors.62 This complex network of feedback between miRNAs and epigenetic pathways appears to form an epigenetics-miRNA regulatory circuit and to organize the whole gene expression profile.63 When this regulatory circuit is disrupted, normal physiological functions are interrupted, contributing to various disease processes or to variability in drug responses, including antiblastic drugs. Myocardial miRNA profiling of murine hearts chronically or acutely exposed to doxorubicin showed downregulation of the miR-30 family through GATA-6.56 The authors concluded that high miR-30 levels are protective against doxorubicin toxicity and correlated this observation with lower ROS generation.56 So far, only 1 research group has reported the upregulation of miR-146a upon doxorubicin treatment in cardiomyocytes, the cells being more resistant to doxorubicin when artificially reducing miR-146a expression in vitro.60 A transgenic murine model overexpressing the caspase recruitment domain (ARC)64 and exposed to doxorubicin treatment showed reduced cardiotoxicity, this effect being mediated by suppression of miR-532-3p.55 In this model, miR-532-3p was found to sensitize cardiomyocytes to doxorubicin-induced mitochondrial fission and apoptosis by targeting ARC.55 Contradictory studies have recently emerged about miR-208a.57,58,65,66 In fact, 1 research group demonstrated the potential of miR-208a silencing against doxorubicin-induced cardiotoxicity in mice.58 The authors administered 20 mg/kg of doxorubicin as a single dose, and after 7 days the mice hearts were harvested and analyzed. As a result, the authors showed a 4-fold increase in miR-208a expression and a pronounced downregulation of GATA4 in the control group. Meanwhile, the other group, pretreated with miR-208a antagomir, showed an attenuation of miR-208a expression and a restoration of GATA4 levels. On the other hand, pretreated mice showed an increase in the expression level of the antiapoptotic gene BCL-2 and decreased apoptosis when compared with the control group.58 In support of the protective role of miR-208a, the study by Vacchi-Suzzi et al.67 showed that the expression level of miR-208a in mice hearts is decreased during doxorubicin treatment. However, contradictory studies have also shown that after administration of 24 mg/kg of doxorubicin the expression level of miR-208b was increased by 8.2-fold in mice hearts, while no change was observed for miR-208a.65 Similarly, another study showed that, after a single administration of a high doxorubicin dose (30 mg/kg) in mice, the circulating level of miR-208a, as well as that of cardiac troponins (cTnI and cTnT), did not change significantly while miR-1, miR-133a/b, and miR-206 were increased.66 In addition, no histopathological changes were observed in mice hearts. Finally, circulating miR-208a was undetected in plasma from breast cancer patients throughout chemotherapy treatment with 4 cumulative doses of 60 mg/m2 doxorubicin.57 Taken together, although the miRNA-based treatment and the more recent epi-miRNAs therapeutics seem to be promising tools to protect the heart against anthracycline-induced cardiotoxicity, we must be very careful once these miRNA are key modulators of gene expression in the heart and its silencing may lead to several cardiac abnormalities (Figure).

Mass spectroscopy has allowed the preclinical identification of promising biomarkers in preclinical models of anthracycline-induced cardiotoxicity.16–20,68–71 The first attempts to use proteomics in anthracycline-induced cardiotoxicity date back to 2004, when Petricoin et al.,72,73 analyzed the mass spectra obtained from more than 200 serum samples of rats treated with doxorubicin or mitoxantone +/- dexrazoxane. Those experiments showed troponin T as a marker of early damage. Remarkably, blood samples from animals treated with docetaxel and adriamycin showed higher expression of proteins involved in energy production pathways, including glycolysis, the Krebs cycle, and the mitochondrial electron transport chain.74 These experiments were performed by Ohyama et al.,74 on heart tissue samples from control rats and rats exposed to docetaxel and adriamycin, and led to the identification of 9 proteins expressed differentially in the control and in the 2 treatment groups. Surprisingly, the expression of glyceraldehyde-3-phosphate dehydrogenase was higher in the group with a lower mortality rate. In addition, Sterba et al.75 analyzed the mass spectra obtained from heart samples of rabbits treated with daunorubicin. The most important alterations were found in mitochondrial proteins involved in oxidative phosphorylation and antioxidant systems.75 The importance of energy metabolism was also confirmed in preclinical models of daunorubicin-induced cardiotoxicity, where proteomics analysis showed alterations in mitochondrial proteins involved in oxidative phosphorylation, energy channeling, and an increased abundance of chaperones and proteins involved in autophagy, membrane repair, and apoptosis.75 Finally, preclinical models of doxorubicin-induced cardiotoxicity showed increased expression of markers for cellular stress (adenosine triphosphate synthase, enolase alpha, alpha B-crystallin, translocation protein 1, and stress induced phosphoprotein 1), and apoptotic/cell damage (p38 alpha, lipocortin, voltage dependent anion selective channel protein 2, creatine kinase, and MTUS1).76
Another interesting and new field of study is the application of metabolomics in the early detection of cardiotoxicity. Unlike proteomics, which aims to assess the entire spectrum of the cell proteins, metabolomics allows the study of small molecules in a biological sample involved in the cell metabolism. Andreadou et al.50,77 examined NMR metabolic profiles in the hearts of rats treated with doxorubicin. They found differences between the control and treatment groups in myocardial levels of acetate and succinate, which were increased in rats exposed to doxorubicin, while levels of branched chain amino acids were decreased.50,77 The authors concluded that acetate and succinate could be useful as biomarkers of cardiotoxicity. Tan et al.78 confirmed the involvement of energetic metabolic reactions in the development of cardiotoxicity. The authors analyzed the metabolic profiles of the hearts of rats treated with doxorubicin using gas chromatography-mass spectrometry metabolomics.78 They found 24 metabolites involved in glycolysis, the citrate cycle and the metabolism of some amino acids and lipids, which were increased in the group treated with doxorubicin.78 More recently, Li et al., 79,80 carried out a metabolomic analysis of plasma samples of mice treated with doxorubicin, using ultra-performance liquid chromatography quadrupole time-of-flight mass spectrometry. The authors identified 39 biomarkers that were able to predict cardiotoxicity earlier than other biochemical analyses and histopathological assessments.80
Taken together, biomarkers identified by the “-omics” approach are considered new potential markers, especially in the scenario of diagnosis and risk stratification of acute coronary syndromes induced by antiblastic drugs, and may be helpful in the early detection of anticancer cardiotoxicity; however, the clinical data available in chemotherapy-induced cardiotoxicity remain insufficient.
CLINICAL OUTLOOK AND FUTURE PROSPECTSIn recent decades, we have witnessed an increase in the effectiveness of cancer therapy, which has led to a decrease in the incidence of cancer. However, the advent of more effective anticancer therapies has led to a higher incidence of cardiotoxicity, with negative impact on quality of life and cardiovascular morbidity and mortality. For the early diagnosis of cardiotoxicity, current guidelines do not recommend evaluation and monitoring of left ventricular dysfunction through the use of different cardiac imaging techniques, because of significant differences in the results across the various techniques. Instead, the guidelines recommend choosing a technology that gives the most accurate possible assessment of cardiac function, and consistent use of the same technique so that controls are comparable and the pulse can be maintained on the impact of anticancer therapy on cardiac function. Because of its safety, wide availability, repeatability and low cost, 2D echocardiography is the most widely performed and standardized procedure in clinical practice and is considered the first-line method to assess ventricular function alterations and to stratify the risk of HF and manage treatments. However, 2D echocardiography requires a significant amount of myocardial damage to unmask cardiotoxicity, precluding every possibility of preventing its development. To diagnose disease at the earliest stages and to predict the risk of its development, it is therefore necessary to integrate echocardiography assessment with other biomarkers that may allow a better stratification in advance and early detection of risk. The “-omics” approach may offer novel and promising tools to detect cardioprotective gene modulators and targeting receptors, with a more robust and predictable approach in cardioprotection and the early detection of cardiotoxicity and individual responses to antineoplastic drugs. This could change the current definition of cardiotoxicity, shifting from a clinical to a subclinical definition, based on earlier, more sensitive and specific biomarkers.
CONFLICTS OF INTERESTNone declared.