



The most important challenge faced by human beings is health. The only way to provide better solutions for health care is innovation, true innovation. The only source of true innovation is research, good research indeed. The pathway from a basic science study to a randomized clinical trial is long and not free of bumps and even landmines. These are all the obstacles and barriers that limit the availability of resources, entangle administrative-regulatory processes, and restrain investigators’ initiatives. There is increasing demand for evidence to guide clinical practice but, paradoxically, biomedical research has become increasingly complex, expensive, and difficult to integrate into clinical care with increased barriers to performing the practical aspects of investigation. We face the challenge of increasing the volume of biomedical research and simultaneously improving the efficiency and output of this research. In this article, we review the main stages and methods of biomedical research, from nonclinical studies with animal and computational models to randomized trials and clinical registries, focusing on their limitations and challenges, but also providing alternative solutions to overcome them. Fortunately, challenges are always opportunities in disguise.
Keywords
Animal research has been the fulcrum of controversy from the outset. In 1543, Andreas Vesalius published De humani corporis fabrica (On the Fabric of the Human Body), and in doing so not only founded modern human anatomy as a scientific discipline but simultaneously placed in question the value of comparative anatomy. He insisted that study of human anatomy required dissection of humans and not close relatives such as apes. Major anatomical findings abounded thereafter but little in the way of animal investigation for medical science. Some 230 years later, the naturalist Stephen Hales described the first measurement of blood pressure. In volume II of Statical Essays,1 he explained how he inserted brass tubes into the crural artery of a restrained, awake mare, and how he then fitted a glass tube into the brass tubes to accommodate the column of rising blood, observing and recording the oscillations in the rising column as a quantitative measure of blood pressure. He did not continue his research, however, focusing his naturalist tendencies on less animate objects, like plants, as his vivisection was greatly criticized. In 1718, his good friend the poet A***lexander Pope, a renowned dog lover, reportedly said of Hales: “He commits most of these barbarities with the thought of their being of use to man. But how do we know that we have a right to kill creatures that we are so little above, such as dogs, for our curiosity, or even for some use to us?”2
It was the nineteenth century that propelled animal work, first through the pioneering efforts of the physiologist Claude Bernard, and then intriguingly through Charles Darwin's theories of evolution. Contrary to prevailing thought, Bernard insisted that all living creatures were bound by the same laws, and in a manner like inanimate matter, and Darwin hypothesized the descent of man from previous forms. Both thought processes suggest that there is much that can be learned from animal physiology of human processes, as the driving forces and laws of nature are preserved even when the anatomy differs. Both Bernard's and Darwin's ideas make the case that fundamental truths of the human condition could well and perhaps even more cleanly be examined in living animal systems. Bernard actively pursued animal work even in an era preceding anesthesia and discovered from this research the digestive properties of the pancreas, the glycogenic function of the liver, and the vasomotor system, creating the concept of milieu intérieur, which Walter Cannon was to term homeostasis. For his science and methods, Bernard is revered to this day, but for his embracing vivisection even his wife and daughter were to vilify him. Darwin was well aware and conflicted in understanding that his theories enabled and in part stimulated the use of animal work.
The advent of anesthesia removed the obvious and readily apparent aspect of the cruelty of absent pain management and enabled greater control over state and reproducibility of effect. Today, the harnessing of the controlled environment of animal work is essential to advance medical therapies including the optimization of life-saving medications such as insulin and virtually every impactful medical device. This is not, however, because animals can model human disease, as there are no models of human disease except in the human. There are no animal models of human disease. Rather, animals are beneficial because they enable what can rarely be performed in human clinical trials–testing in a precise framework of hypotheses regarding mechanism of action. Just as efficacy and safety can be hinted at in animals but only proved definitively in humans, the reverse is true of mechanisms. Hypotheses about mode of action can rarely be proved in the extraordinarily variable human conditions; they require controlled environments where many conditions can be held constant and ideas can be tested, ie, animals. Animal experimentation is critical because it is in these living beings that physiologic concepts can be validated in a manner that could not otherwise be tested. However, the value of such study necessitates that it be performed only with absolute commitment to respect for living beings and precision of trial execution. Inappropriate models, eg, ones where comparisons cannot be made, where the physiology is fundamentally different, inappropriate control of animal experiments, where poor or improper attention to animal needs is not only cruel and unethical but inevitably taints the results because distress states are uncontrolled, and where there is no human follow-up, all invalidate their value. In fact, once an animal trial is performed and confirmed, human trials must be performed to define safety and effect compared with predicate forms. This is an important, not a passing, caveat. Just as one should seriously worry when clinical trials are performed without the mechanistic support from animal work, so too should one consider that successful animal work that is not followed by clinical trials is wasteful and disrespectful. Thus, animal work should not be performed when there is no hope of a translational impact of its own or anticipation of ultimate clinical validation.
Here then are the modern dilemmas–what is to be done when animal work is so far from the human experience as to require creating devices unique to the animal, and then how should we consider the current rush to clinical trials before complete comprehension of effect? To a large extent, these are different sides of the same coin. The testing, for example, of percutaneous heart valves is significantly limited by the stark difference in the anatomy of the aortic arch between quadrupedal sheep or swine and upright, bipedal man. Valves that are to be considered for use in people are exceedingly difficult to insert through the steeple-like aorta of the former, limiting the usefulness of the animal model. Insistence on performing animal work with these devices might require the creation of devices that could only be used in animals. Such a solution is unnecessary and distracting. Distraction resides in the pursuit of an irrelevant model but the unnecessary aspect is because the animal is not the only nonclinical model and its inaccessibility does not mean that one cannot validate basic operational and mechanistic issues before human use. There are a multitude of even more appropriate models available on the bench and in silico that can provide conceptual insight before human trials. Eschewing these preclinical alternatives in favor of premature clinical testing is a violation of scientific method.
At the same time, the rush to perform trials has created a countervailing set of issues that are potentially equally damaging. The endovascular implant experience has fueled cardiovascular intervention in the most extraordinary way, propelling vascular biology and medical science in parallel with innovative technology. The hallmark of this experience, though, is deep attention to precise multimodal and multidimensional preclinical evaluation before pivotal human trials. The critical preclinical evaluation of bare-metal stents preceded clinical trials and Food and Drug Administration (FDA) consideration and approval by 5 years, and the first definitive publication of the promise and mode of action of drug-eluting stents came 5 years before FDA approval of these devices.3,4 When issues arose with these devices, there was an abundance of animal, benchtop, and computational work to direct further clinical evaluation. In contrast, the same cannot be said for renal denervation and, to a lesser extent, for bioerodible scaffolds. The Simplicity III HTN trial was performed with rigor and care, and unequivocally demonstrated no effect of radiofrequency renal denervation compared with sham controls.5 Because of the strength of this trial, most renal denervation development programs have ground to a halt and yet initial trial enrollment on the heels of promising early clinical trials was rushed before definitive animal work could be published.6,7 Only in retrospect do we now realize how the clinical approach would have been changed had we known beforehand of the animal trial data that emerged after the clinical publications. Perhaps if these articles had appeared first, we could not only have designed the trial differently but also responded in a less binary, on-then-off manner to the failure to demonstrate benefit. This is also true of erodible scaffolds. Early publications in the field of preclinical evaluation of implants suggested that erodible systems required far longer observations in animals than similar devices in durable materials before the start of clinical trials to account for the anticipated time to material erosion and clearance.8,9 This was not actually adhered to and, soon after repeated clinical data demonstrated that one erodible scaffold had increased rates of thrombosis, the FDA issued a warning regarding the use of this device. Only now, after definitive clinical demonstration of a safety issue, thrombosis, are we realizing that performance follows what could have been predicted from animal and computer models of material erosion. The erodible scaffold, with twice as much material surface area and twice the strut thickness, exhibits twice the rate of early thrombosis. Moreover, rates of thrombosis continue to diverge for the erodible and durable material and peak again late, coinciding with the inflammatory conditions necessary for and accompanied by material erosion. Once again, this illustrates a definitive clinical result and profound on-then-off reaction to faith in the technology but before achieving a full understanding of the engineering and science of the device. We fear that definitive scientific and engineering papers will not be published, or read, if presented, with the care and attention they would have garnered had they been published first.
The problem when clinical trials do not wait for animal work–premature introduction of a technology onto the market–not only exposes participants to incompletely vetted technology, but, as has happened with renal denervation and likely erodible scaffolds, also prematurely eliminates a potentially promising set of interventions before they can be definitively determined to be flawed. It is only the most intrepid or least informed who will continue to support a technology tainted by failure, even when failure arises from flawed trial placement in the development cycle. Here we must distinguish not just a well-executed from a poorly-executed trial, but also trials that should and should not have been performed.
What then is the balance? We must first and foremost acknowledge that therapeutic design and development is about balance–one must seek to understand to the extent possible and to proceed to the clinical domain when risk mitigation can no longer be extended. One must also concede that there are no absolutes. There is no such thing as absolute safety–no device or drug can be entirely effective and fully safe; the idea of complete efficacy bedevils the notion of absolute safety. There is a fine line between the most potent therapeutic effect and the emergence of undesired side effects. At the same time, there is little to be gained from a “let's try it and see what happens” approach. Such an approach is most often doomed to failure, and even in success, as in failure, there is no understanding of why the result was observed, and therefore little chance for rational rectification. As such, an integrated approach should seek first to formulate and then to test a hypothesis of action in as many domains as possible, including multimodal benchtop testing, in silico modeling, and then use in animal models under the most controlled environments, in escalating levels of complexity.
There is one important comment in closing and it is that clinical trials are best performed with as much conceptual support and mechanistic understanding as possible but do not need to wait for completion of all possible nonclinical work. This is not the point of nonclinical work, ie, to proceed to clinical trials. Rather, nonclinical work motivates progression to clinical trials as the next logical step in providing a transformational therapy, diagnosis, or disease reclassification. In fact, we prefer the term nonclinical to preclinical to avoid promoting the idea that the terminal phase of investigation is clinical. There is a burning need for nonclinical investigation to proceed in parallel with and well after clinical trials. Clinical observations like erodible scaffold thrombosis and renal denervation inefficacy beg further nonclinical investigation. It is precisely when clinical observations defy expectations that added investigation in the nonclinical domain are warranted.
In short, device development cannot be complete without animal model systems, as it is only in this nonclinical domain that hypotheses can be tested and observations can be explained. The clinical environment proves clinical benefit and defines the window of safety, but it is nonclinical investigations that are essential to explaining how systems work and why, and the effects of changes in environment, stresses, and loads. We remain dependent and indebted to these systems and need to honor, respect, and consider them with the same scientific, ethical, and technical benchmarks as those used in clinical trials.
RANDOMIZED CLINICAL TRIALSThe Foundation for Evidence-based MedicineSince the first randomized clinical trial (RCT) in 1948 (Medical Research Council Streptomycin Trial), RCTs have reshaped medical knowledge and practice, reducing bias and enhancing the accuracy of clinical experimentation. Randomized clinical trials have achieved the status of gold standard for evidence-based medicine. These trials have revolutionized medical research and improved the quality of health care by clarifying the benefits and drawbacks of countless interventions.10 They provide the highest level of evidence to support recommendations in guidelines and are required by regulatory agencies for drug and device approval, although with variable degrees of scrutiny.
The importance of RCT-based evidence is clearly stated in a recent investigation aiming to assess the agreement of treatment effects on mortality from registries and subsequent randomized trials. The evaluation included 16 eligible observational studies, and 36 subsequently published RCT investigating the same clinical questions.11 For 5 (31%) of the clinical questions, the direction of treatment effects differed between observational studies and trials. Confidence intervals in 9 (56%) observational studies did not include the RCT effect estimate. Overall, observational studies showed significantly more favorable mortality estimates (by 31%) than subsequent trials. Therefore, registries could provide different answers from subsequent RCT on the same clinical questions and may substantially overestimate treatment effects.
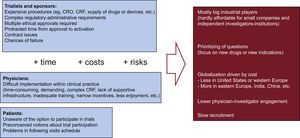
It is clear that large randomized trials might still be needed to address critically important clinical questions for patient-relevant outcomes. However, although RCT in cardiovascular medicine have transformed care, in a review of 16 disease-specific, diagnostic, and interventional guidelines, only 12% of recommendations were level A (with heart failure the highest at 26% and valvular disease the lowest at 0.3%), and 48% of recommendations were level C.12 Of course, if no randomized trials exist, clinicians can still act on the results of observational studies, but they should consider that treatment effects could be more uncertain. Nevertheless, the past 7 decades also bear witness to many limitations of this “gold standard” (Table 1, Figure 1).
Randomized Controlled Clinical Trials
| Strengths |
| Correctly designed and with adequate power, they are the gold standard |
| Eliminate bias and confounding |
| Establish causality between intervention and outcome |
| Challenges |
| Complex regulatory requirements |
| For-profit contract research organizations |
| Multiple ethics approvals |
| Many unknowns for power calculation |
| Not always sized enough, underpowered for hard events (ie, death), powered for composite “hard+soft” endpoints (ie, death+readmission) |
| Prescreening is ineffective and unpredictable |
| Slow enrollment (early interruption) |
| Highly selected populations due to exclusion criteria (exclusion of high-risk populations and exclusion of off-label indications) |
| Limited applicability |
| Often select specialized study centers |
| Often surrogate endpoints |
| Limited follow-up time |
| Long time to plan and complete |
| Very expensive |
| Often sponsored by industry-only (interest in expensive, novel patented therapies) |
| Frequently conducted postmarketing (results available a long time after approval) |
| Reporting can be suboptimal (late or no publication at all) |

Even as RCT have become standard in pharmaceutical research, clinical researchers have struggled in the last few decades to apply them to other areas of medicine such as surgery, for example. As more surgical RCTs appeared in the 1960s and 1970, surgeons increasingly recognized their limitations: each patient had unique pathological findings, each surgeon had different skills, and each operation involved countless choices about anesthesia, premedication, surgical approach, instrumentation, and postoperative care, all of which defied the standardization that clinical trials required. Sham controls could not be used for major operations, which limited opportunities for blinded trials.13
Even well-conducted RCT have sometimes failed to influence medical practice. The reasons are multiple, from market pressure to inertia and skepticism. In 2002, The ALLHAT trial revealed that generic thiazide diuretics were as effective as newer, expensive calcium-channel blockers and angiotensin converting-enzyme inhibitors in treating hypertension.14 However, sales of the newer antihypertensives grew faster than those of diuretics.
The use of intra-aortic counterpulsation balloon pump has not decreased as expected after the landmark trial showing no benefits on mortality.15
On the other hand, some RCT results have been accepted as fact but have later proved to lack external validity. Social and ethical concerns have also challenged the legitimacy of some RCT. Some advocate for more flexible approaches to clinical research, including the use of surrogate endpoints, conditional regulatory approvals, and parallel tracks to provide access to drugs outside of trials. However, critics worried that the loosened standards undermined scientific rigor and encouraged a risky deregulatory agenda championed by the drug industry.
One long-standing concern has been the discrepancy between the time frame of RCT and the fast pace of innovation. Just when we have accumulated enough data over a sufficient time period, we find that a specific surgical/interventional technique has improved or medical therapy has changed, or both, and conclusions no longer apply.
RCT Are, However, “Gold-priced”RCT have become increasingly large, complex, and expensive, which may threaten their very existence.16 Currently, a single phase 3 RCT could cost up to $30 million or more. A large trial with 14 000 patients recruited in 300 centers cost $300 million dollars.17 However, the cost is not the same across the world, with the United States being the most expensive country, while the cost is 50% lower in Germany, 39% lower in Poland and 36% less in India, relative to the United States. Consequently, countries now compete to convince the pharmaceutical industry and contract research organizations that their regulatory, clinical, and public health profiles provide ideal trial conditions, even when the products being tested are unlikely to be made available to local populations after trial completion.18
As RCT developed into high-cost, high-value marketing tools, a clinical trials industry burgeoned. Having emerged in the late 1970s, contract research organizations have become a highly profitable industry.19
Pitfalls in RCT Design and ReportingThe design and reporting of RCT is not uncommonly suboptimal. Of 96 346 studies registered in ClinicalTrials.gov between 2007 and 2010, the majority were small and with heterogeneous reporting of methods.20 Among 13 327 trials registered between 2008 and 2013, only 13% reported results within 12 months of completion.21 Among 244 extramural trials funded by the National Heart, Lung, and Blood Institute and completed between 2000 and 2011, only 64% had been published by 2012 and the median time to publication was 25 months.22
The results of the REVIVE trial evaluating the calcium sensitizer, levosimendan, in patients with acute decompensated heart failure were published more than 7 years after the trial was completed and yet the drug had been available in over 40 countries.23
Guidelines have been developed to improve the quality of RCT reporting. The implementation and endorsement of some of these guidelines, such as the CONSORT Statement, have been empirically shown to improve the quality of reporting.24–26
Barriers and Opportunities for RCTAmong the challenges and barriers for running RCT (Figure 1), cost reigns supreme. The extremely high costs imposed make the large trials only affordable to big companies interested in their new drugs or devices. Prioritization of research questions by companies is different from the priorities of society in general, which prioritize comparison of commonly used therapies.
The complex administrative and regulatory procedures demand a very long time and the risks of failure have to be assumed by sponsors. These multiple small obstacles are especially cumbersome in the particular case of investigator-initiated trials.
On the other hand, physicians face many problems for active participation (mostly from their home institutions). Last but not least, patients are either unaware of the option to participate or, among those who are aware, many choose not to participate, because of preconceived notions about safety or privacy.
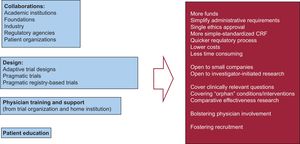
Fortunately, there are alternatives to overcome most, if not all, of these barriers (Figure 2). The most important is to promote active collaborations between academic and health institutions, industry, disease-focused foundations, patient groups, and administrative-government agencies at the national and supranational level. In recent years, large trials in cardiovascular medicine, such as the DAPT trial, have been funded by collaboration between academic institutions and industry.27 These collaborations have evolved to address the therapeutic challenges of the so-called orphan diseases, such as cystic fibrosis or type I diabetes.28 Coordinated prioritization of research needs can be seen in the increasing interest in comparative effectiveness research by the health administrations in some countries.

Other important alternatives rely on adaptive trial designs, physician support at home institutions and patient education programs. With regard to the adaptive design approach, this may be applied either to exploratory clinical trials (finding safe and effective doses or with dose-response modeling) or confirmatory trials (making prospectively planned changes to the future course of an ongoing trial on the basis of an analysis of accumulating data from the trial itself). There are examples of trials undergoing sample size re-estimation or changing the primary endpoint.29,30
Another alternative for adaptive design comes from the use of biomarker-driven population enrichment designs. There is a potential for predictive biomarkers to identify patients who are likely to benefit from targeted therapies and to thereby increase the success rate of confirmatory clinical trials. In this design, all participants undergo randomization regardless of biomarker status but with the use of an interim analysis to identify whether biomarker-positive patients benefit differentially from the tested agent compared with biomarker-negative patients. If only the biomarker-positive patients are benefiting, then further enrollment in the biomarker-negative subgroup would be halted. The final statistical analysis of the data would be based on data from the 2 stages with the use of closed testing and conditional error rate methods to prevent inflation of the type I error.31
Meta-analysesIn recent years, we have witnessed an exponential increase in the number of meta-analyses published. No topic in cardiovascular therapeutics lacks a corresponding meta-analysis. In fact, in any given year, several pooled analyses addressing the very same topic are published.
The benefit of this approach is the pooling of studies (trials and observational), leading to a higher statistical power and more robust point estimates than is possible from the measure derived from any individual study. However, when performing a meta-analysis, the investigator has to make choices that affect the results, such as the search criteria, how to deal with incomplete data, or what statistical methods should be used.
It is important to know the limitations of this approach. A meta-analysis of several small studies does not predict the results of a single large study. A good meta-analysis cannot correct for poor design and/or bias in the original studies. Only methodologically sound studies should be included in a meta-analysis (best-evidence synthesis).32
Another potential pitfall comes from the publication bias or “file drawer” problem. This implies reliance on published studies, which may create exaggerated outcomes due to publication bias, as studies that show negative results or insignificant results are less likely to be published.33 In addition, disclosures of interest are not always fully reported and there is a potential problem derived from agenda-driven bias. Other weaknesses derive from the statistical methodology because the most accurate statistically method for analyzing pooled results remains to be determined. In this regard, there is a mounting criticism of the popular random effects model.
Guidelines are provided by the PRISMA statement, which establishes an evidence-based minimum set of items for reporting in systematic reviews and meta-analyses.34
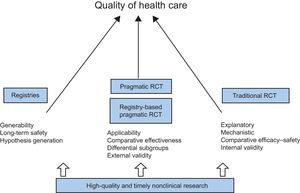
Pragmatic Randomized TrialsA distinction could be made between RCT according to their main focus. Explanatory or mechanistic trials are focused on the question, “Can this intervention work under ideal conditions?” whereas pragmatic trials seek to answer the question, “Does this intervention work under usual conditions?”35
A tool has been developed that establishes a set of criteria to help researchers determine how pragmatic or explanatory their trial is. The tool was originally called PRECIS (Pragmatic-Explanatory Continuum Indicator Summary) and now has a new version, PRECIS-2.36,37 In addition, a pragmatic extension to the CONSORT statement has been proposed.38
Pragmatic trials are designed to show the “real-world” effectiveness of interventions in broad patient groups and to identify the subgroups for whom the innovation will provide the greatest net benefit. Therefore, these are characterized by large sample sizes; representative populations and generalizable and relevant outcomes, efficient use of existing resources, simplified operations (limited monitoring, safety reporting, trial-specific assessments, and regulatory and compliance documentation), baseline and, if possible, outcome data collection embedded in routine care settings or using telephone or automated follow-up, leveraging of electronic health records (EHR) and registries, and simplified case report and informed consent forms.39–41
The key dimensions that define a pragmatic trial are focused on the recruitment of patients and investigators, the type of interventions and their delivery, the type of follow-up, and the type and determination of outcomes.
In pragmatic trials, participants should be similar to patients who would receive the intervention if it became usual care. Inclusion and exclusion criteria should be minimized and the number and complexity of study visits and procedures optimized. In some contexts, it is even possible to waive informed consent. This is the case of cluster randomization, which involves groups of patients (in the same health care facility) who are randomly assigned to the same intervention.42
A related approach is the cohort multiple-randomized design, in which a cohort of participants is recruited and consent is obtained for follow-up and possible recruitment into trials of new treatments vs standard care.43
With regards to investigators, pragmatic trials should include a variety of investigators with a representative mix of experience appropriate to the intervention under study. If heterogeneity in responses to the intervention is likely, a trial must be large enough to permit an understanding of that heterogeneity. The development of clinical networks, the establishment of disease-specific research communities, and providing credit to health professionals for research are definitely helpful.
The delivery of the intervention needs to be as close as possible to normal practice. This means that pragmatic trials are commonly unmasked. Therefore, reporting of nonserious adverse events, the reasons for treatment discontinuation, and several patient-reported outcomes are subject to greater degrees of bias, affecting the quality of the trial. To minimize biases, it is important to focus outcomes on major events (ie, death or emergency hospital admissions).
Follow-up should ideally be accomplished through the use of EHR. This strategy is only feasible in health care systems with reliable, standardized, and accessible EHR that capture the events of interest. The recruitment of patients who are already enrolled in disease-specific or intervention-specific registries provides an efficient and low-cost opportunity for conducting pragmatic trials, such as the TASTE trial from the Swedish cardiovascular registry.44 Many trials have been conducted under this pragmatic focus, but most have not used registries that have the added benefit of baseline and in some cases outcome data being embedded in routine clinical care or accessible by automatic linking of data sources.45
In relation to outcomes, pragmatic designs could portend more limitations to assess objectives requiring procedures that are not part of routine practice. The presumed lack of need for events adjudication in pragmatic trials is debatable, since events adjudication is more a quality concern than a pragmatic issue.
The discrepancy between mechanistic and pragmatic trials could be expressed, in a somewhat simplistic way, as greater internal validity of mechanistic studies vs enhanced external validity of pragmatic trials. It is important to keep in mind that the features of pragmatic trials that support the generalizability, or applicability, of their results, such as the inclusion of heterogeneous patient populations, lack of blinding, absence of a placebo group, or suboptimal treatment adherence, may also reduce sensitivity and limit the interpretation of the results. Not all clinical questions can be answered by a pragmatic design; thus, this approach must be applied whenever feasible and when not compromising trial quality and the ability to answer the clinical question of interest.41
Pragmatic Registry-based Randomized TrialsAs mentioned previously, EHR and clinical cardiovascular quality registries are providing opportunities for pragmatic and registry-based prospective RCT. Simplified regulatory, ethics, and consent procedures; recruitment integrated into “real-world” care; and simplified or automated baseline and outcome collection allow assessment of study power and feasibility, fast and efficient recruitment, delivery of generalizable findings at low cost, and potentially evidence-based and novel use of generic drugs with low costs to society.46
The Swedish SWEDEHEART registry has been the platform for the first registry-based RCT (TASTE trial) and more recently the iFR-SWEDEHEART trial.44,47 Likewise, the American College of Cardiology has collaborated with member investigators (and with support from the National Heart, Lung, and Blood Institute) to pilot the use of the CathPCI Registry as the data collection backbone of a RCT comparing radial and femoral access in women undergoing cardiac catheterization.48
The unique features of the Swedish registry will be discussed in the following section of this review, but it is critical to facilitate the development of this sort of RCT. The possibility of extracting both baseline and outcome data from quality registries linked to administrative databases is key for accomplishing these trials.
Pragmatic registry-based RCT offer benefits and potential limitations of their own (Table 2). The most relevant advantages are related to the ability to recruit much larger representative samples, in a shorter time and with significantly lower costs. In addition, given the minimized monitoring, the most important limitations are related to data quality, uniformity in events adjudication, and privacy concerns.
Pragmatic (Registry-based) Randomized Clinical Trials
| Strengths |
| Randomized “real-world” evidence |
| Simplified regulatory process |
| Nonprofit academic research organizations |
| Single ethics approval |
| Sample size powered for relevant events, know eligible sample and event rates |
| Prescreening is automated, efficient, and predictable |
| Unselected patient populations–generalizable: efficacy and effectiveness |
| Large number of events, allowing identification of rare events |
| Automatic outcomes assessment |
| Less expensive |
| Promote adoption of evidence into practice |
| Evaluate new use of existing drugs/devices |
| Weaknesses |
| Data quality |
| Events adjudication |
| Blinding for interventions |
| Minimized monitoring with implications for safety in trial participants |
| Privacy in these large patient databases |
| Balance efficacy vs effectiveness |
| Assess effects on patient-reported outcome measures such as quality of life |
| Performance at a multinational level |
Although registry-based RCT are much cheaper than traditional RCT, they still entail considerable expense, usually beyond the possibilities of institutional or investigator grants. It is remarkably important for stakeholders, including public regulatory and funding agencies, to recognize the need for trial reform and the advisability of funding pragmatic trials. Now is the time for both industry and public funders to leverage these emerging new ways of conducting RCT based on their efficiency and inexpensiveness, leading to new treatments for patients combined with savings for shareholders and the public.49–53
CLINICAL REGISTRIESA clinical registry is an observational database focused on a clinical condition, therapy, or population. Data are collected systematically for specified scientific, clinical, or policy purposes. There are no mandated approaches to treatment in clinical registries and they have broad inclusion criteria with few exclusion criteria. The focus of clinical registries is on capturing data that reflect “real-world” clinical practice in large, representative patient populations.54
Registries can be classified on the basis of the characteristics of the population enrolled or by purpose (quality measurement, research, or multiple). These may be either prospective or retrospective in design and are designed and executed by many types of entities (researchers, professional societies, nonprofit organizations, government agencies and industry). Clinical registries capture data under standardized definitions and standards. Although data usually comes from medical record abstraction and case report forms, some registries extract data directly from generalized EHR.
Performance and Quality of CareOver the past few years, there has been an increasing emphasis on measuring and improving the quality and efficiency of medical care and subsequently there has been a proliferation of clinical registries designed to evaluate care performance and outcomes in “real-world” settings. Clinical registries represent a key element in measuring the outcomes of care processes, providing actionable feedback to health professionals, and improving quality of care. Assessment of health care delivery is increasingly done by means of registries including the following55:
- •
Assessing health care effectiveness and safety.
- •
Measuring appropriateness of care and disparities in its delivery.
- •
Measuring the effectiveness of quality improvement.
- •
Evaluating factors that influence prognosis and quality of life.
- •
Improving clinical outcomes, patient care experience, and patient-reported outcomes.
A recently published report provides data for 2014 from 4 National Cardiovascular Data Registry hospital quality programs in the United States, showing trends in cardiovascular care.56
All agents involved in health care benefit from registries:
- •
For health professionals, registries improve their quality of care and patients’ clinical outcomes and enable personalized medicine.
- •
For payers, registries evaluate quality and the cost of care (cost-effectiveness).
- •
For industry, registries demonstrate the value of products, and bring new drugs, devices, and services to the market more quickly, with a higher likelihood of clinical and economic value.
- •
For patients, registries mean a more active participation in the health management process and allow an ongoing dynamic interaction with health providers, bolstering treatment adherence and promoting healthy behavior.
Undoubtedly, the gold standard for evidence-based medicine generation is the RCT. Cardiology is truly an evidence-based specialty, yet only a minority of practice guideline recommendations is actually supported by the highest quality level of evidence.12 For many clinical questions, no RCT has been properly done and most probably will never be done, mainly due to ethical and economic considerations. Interventions not related to novel patented drugs, devices and services (orphan interventions) are scarcely evaluated in RCT due to a lack of funding interest.
As discussed previously, although randomized trials are considered the gold standard for comparative effectiveness, they have some limitations (Table 1). Mainly, RCT are becoming increasingly more expensive and recruitment is generally restricted, resulting in problematic generalizability.
Restrictive Inclusion and Applicability in RCTIt is estimated that less than one third of heart failure patients in clinical practice would qualify for inclusion in RCT and nearly two thirds of the participants in the Euro Heart Survey on Coronary Revascularization would have been ineligible for participation in RCT comparing percutaneous coronary intervention with coronary artery bypass grafting.57–59 Patients in clinical practice were older and more likely to have comorbid conditions. Although RCT showed no difference between percutaneous coronary intervention and coronary artery bypass grafting in outcomes, in the trial-ineligible patients, a clear 1-year survival benefit was reported for percutaneous coronary intervention over coronary artery bypass grafting.59 The conflicting results of an analysis of a New York registry might be explained by this contrast between trial-eligible and trial-ineligible patients.60
Prospective registries are used to assess whether RCT findings in selected populations can be transferred to the overall clinical population. If higher-risk patients are not adequately represented in RCT, registries play an important role in validating trial findings in groups that are excluded or underrepresented. The analysis of these subgroups through registries is of paramount importance, given the increased rate of complications in these patients and the potential for therapies to cause harm. In this regard, patients with advanced age are systematically excluded or minimally represented in RCT.61
Limited Power and Limited Time Scope in RCTAdverse events that occur very late and/or with a low incidence can be easily missed by RCT due to insufficient sample size or limited follow-up time. This was the case of the very late drug-eluting stent thrombosis events and the trials conducted with the first generation of drug-eluting stents.62 Registries with larger populations and longer follow-up periods identified the increased associated risk.63,64
This is particularly relevant with new medications, which have the potential for rare adverse effects. After marketing surveillance, it is crucial to establish the safety of new therapies that are approved under relatively underpowered trials with a limited follow-up time. Moreover, registries can raise safety concerns that have not been detected in previous trials, not because of their limitations but because these simply have not been properly conducted or are not yet finished, or because of insufficient time since device approval. This is the case of the risk of thrombosis with bioresorbable vascular scaffolds.65
Limitations of Registries for the Generation of Medical EvidenceThe strengths and weaknesses of registries are shown in Table 3. Prospective registries are not randomized, and findings on treatment efficacy should therefore be taken cautiously. The presence of small imbalances in unmeasured confounders that have a strong relationship with outcomes can have a large confounding impact on the relationship between treatment and outcomes. As an example of the potential impact of unforeseeable confounders, the initial analysis of the 2003 to 2004 SCAAR cohort indicated increased mortality rates associated with drug-eluting stents compared with bare-metal stents, which was reversed in the analysis of the 2003 to 2006 cohort.66,67 The reversal was attributed to an improved balance in lesion and stent characteristics between the 2 groups in the latter study.
Clinical Registries
| Strengths |
| Unselected patient populations–generalizable |
| Very large populations |
| Epidemiological characterization |
| Large number of events–allows identification of rare events |
| Efficient |
| Inexpensive |
| Possibility of longitudinal follow-up |
| Possible dynamic design features to be implemented while running |
| Comparative outcomes, hypothesis generating |
| Weaknesses |
| No comparative effectiveness, very limited for comparative outcomes research |
| Impossibility of adjusting for confounding factors, despite advanced statistical models |
| No blinding for interventions |
| Nonconsecutive strict recruitment guaranteed (inclusion bias) |
| Data quality variable and questionable |
| No uniform and no centralized events adjudication process |
| Prejudices or preconceptions about treatment effects may bias events reporting |
| Limited or no audits |
Bias may be attenuated, although never fully eliminated, by design and/or analysis. In relation to design, the use of matching criteria for inclusion/exclusion and the use of paired availability (chronological or geographical) may be implemented. In the analysis phase, the use of covariate adjustments, matching, and propensity scoring, along with other currently available statistical methods may help.68,69
In accordance with the aforementioned limitations of the registries, one of the best-established roles of registries is hypothesis generation, suggesting RCT that may confirm or disprove the registries’ findings. As an example of the latter, the use of estrogen therapy in women was beneficial in registries but not in an appropriate trial with respect to cardiovascular outcomes.70,71
The use of large datasets may be used to suggest RCT participation on an individual basis through factor analysis methods, yielding those factors that make patients comparable in terms of prognosis.
As discussed previously in this review, clinical registries can be used as a platform for developing randomized trials and performing comparative effectiveness research, potentially accomplishing the dual objectives of decreasing trial costs while simultaneously increasing the generalizability of the results.
Keys for a Successful RegistryProperly measuring clinical outcomes requires standardized clinical nomenclature, uniform standards for defining and collecting data elements, strategies to adjust for patient complexity, techniques to verify the completeness and accuracy of data, and longitudinal data collection.72,73 The key features for a high-quality registry and upcoming developments can be delimited in a step-by-step fashion (Table 4).74
Key Features of a Successful Registry
| Steering committee and principal investigator designated from the beginning |
| Proper ethics review procedures |
| Standardized definitions for data collection and events reporting |
| Randomized selection of centers (ideally, 100% participation) |
| Consecutive enrollment of patients for representativity |
| Electronic data capture with clear, simple explanations of definitions and instructions for participants, and plausibility controls to highlight incorrectly entered data |
| Integrated tools for rapid feedback to participating institutions |
| Centralized data compilation and statistical analysis, performed by professional statisticians |
| Audit of at least a small group of randomly selected centers |
| Reporting of all collected data, with conclusions appropriate to study the design |
| Transparent reporting of investigators and funding sources in all publications |
| Future developments: |
| • Increased interoperability, enabling governance/workflow management |
| • Implementation of robust machine learning procedures |
| • Internet of things: less dependence on manual data entry, new data sources, patient wearable self-tracking devices, digital ecosystems |
| • Multidimensional phenotyping of patients: medical geographic information system, from demography to overall “-omics” |
The growth of EHR is both a challenge and an opportunity with regard to the potential use of clinical registries. As hospitals increasingly use EHR, it is preferable to automatically extract some registry data elements directly from EHR rather than having to enter them manually. Ultimately this strategy saves time and money. The interplay between EHR and clinical registries is a win-win situation. Electronic health records and administrative data could facilitate longitudinal patient follow-up and capture nonclinical outcomes, such as resource use. Registries, in turn, could bring the discipline of common data models and of systematic data definitions and data quality to EHR.
However, there are important concerns55:
- 1.
Data entry into dedicated clinical registries is accomplished by trained staff who abstract information from the clinical record in accordance with specific definitions. On the other hand, data in EHR are captured in the process of patient care by various health professionals for purposes other than analysis and reporting.
- 2.
Automatic data extraction is feasible for some data, such as laboratory results and demographics but extraction from EHR of more detailed elements in clinical registries will require the personnel administering these 2 data sources to work collaboratively on data element definitions or to blend electronic data abstraction with manual abstraction of nonextractable concepts.
- 3.
Registries generally have processes in place to ensure data quality, but in contrast, EHR data are generally not subject to formal audit.
The main barriers come from the need for dedicated staff, software, data warehouses, and analytical centers. Execution of registries frequently relies on committed volunteer physician leaders. It is crucial to get support either from public institutions (government agencies, hospitals, and other public health institutions) or industry. For this purpose, it is of paramount importance to see these expenditures as investments in the future.
Widespread implementation of registries requires the standardization of data element definitions across all different registries in a particular field. Data quality standards are required but these are not always available, or if so, they are not widely and properly implemented.
The limited time frame for data collection (ie, only the hospitalization period) and the specific nature of the outcomes (ie, only death) are sources of pitfalls and flaws. In this regard, data extraction from EHR could facilitate longitudinal patient tracking and capture of a wide spectrum of outcomes.
Regular external audit programs are essential to verify high accuracy in data, these being in accordance with the accepted standards.75,76
Overcoming Barriers and Limitations in RegistriesStandardized methodologies are crucial to the quality of registry data, and facilitate comparisons between the findings of different registries. This is of upmost importance when a large number of centers and countries are involved in a single registry. In Europe, the CARDS (Cardiology Audit and Registration Data Standards) have been developed.77
In the United States, a report from the Data Standards Workgroup of the National Cardiovascular Research Infrastructure Project provided standardized cardiovascular data for clinical registries.78
At a time of extreme cost containment, the role of registries in clinical care improvement and the subsequent financial benefit should be emphasized. Clinical registries should interface with EHR when possible, minimizing manual data entry, reducing effort and cost. Both newly developing and more mature registries should share data and learn from one other.
Large data management and sophisticated powerful statistical methods will definitely be helpful to enhance the potential for extraction of meaningful and valid knowledge from registries overcoming to some extent the limitations of nonrandomized studies. Machine learning techniques, deep learning, and neural networks may help to predict disease progression and specific treatment effects for individual patients based on rich datasets, complementing other levels of research. Machine learning allows the development of algorithms that use readily available clinical data to identify patients with a specific condition (disease or stage of disease).79–81
Clinical registries must adapt to collect patient-centered performance measures. The amazing development of self-tracking wearable technology for health purposes provides a great opportunity. The development of novel electronic tools for collecting standardized, patient-reported outcomes will be invaluable.82,83
Patient-powered RegistriesTraditionally, registries have been researcher-generated. Patient-powered registries are similar in many ways to researcher-generated patient registries in definition, purpose, and features but in patient-powered patient registries, patients and family members “power” the registry by managing or controlling the data collection, the research agenda for the data, and/or the dissemination of the research.
Patient-generated patient registries in particular have been criticized on several levels and improvements are warranted on data standardization and quality, patient education to enhance their ability to participate, and aspects regarding competition for patients and caregivers.
Without question, patient-powered patient registries and networks are a rapidly evolving contributor to research, and particularly to research that focuses on direct improvements in practice. These entities blur traditional boundaries, breaking down the barrier of patient, family, and advocate involvement and control in research, translation, and dissemination. A clear movement has emerged to connect individual patient organizations and single-condition patient registries into broader networks that unify, standardize, and optimize data collection and research generation processes.84,85
Euro Heart SurveysThe European Society of Cardiology designed the Euro Heart Survey program to assess the applicability of evidence-based medicine, the application of guidelines in clinical practice, and the outcomes of different patient management strategies.85 As mentioned above, data standards (CARDS) have been developed to encourage uniform data collection across countries within Europe.77,86
The Inspirational Swedish ExperienceThe SWEDEHEART cardiovascular registry started in 2009 as a merger of 4 previously existing registries, RIKS-HIA: Acute coronary care registry (1995), SEPHIA: Secondary prevention registry (2005), SCAAR: Angiography and percutaneous coronary intervention registry (1998) and the Swedish Heart Surgery registry (1992).87
Despite the noncompulsory nature, the registry covers the whole activity in the country and is linked to mandatory governmental registries such as the national patient registry, cause of death registry, and dispensed drug registry. The coordinators elaborate and release an annual registry of activities.88
In the words of their own leaders, the keys for success have been the following: a) initiation by cardiologists and propulsion by national and local enthusiasts; b) use of simple registration process; c) inclusion of highly motivated users with direct access to study reports and statistics; d) provision of immediate benefit at the local unit on-line reports; e) possibility of open comparison of hospital performances; f) conceptualization of SCAAR as not just a registry but as a tool to provides clinical information that impacts on quality of care; g) flexibility, users can influence the contents; and h) a high degree of transparency.
Regarding the derived investigational procedures, the functional aspects that have been crucial are the following:
- •
Each hospital owns its own data; participation in national reports and scientific databases is voluntary.
- •
Research projects based on the national database must be approved by the SWEDEHEART steering group.
- •
All projects must be approved by an ethics committee.
- •
Any database is deidentified before reaching the scientist.
- •
Statistical analyses are often done in collaboration with an epidemiologist/biostatistician from a national competence center.
A large number of publications addressing multiple topics in cardiovascular care have been derived from the registry. Only in 2015, 57 publications were generated from the registry.
The registry is facing recent and future developments, such as the following: a) a randomization module for prospective randomized registry-based trials; b) integration with patient EHR; c) enablement of direct reporting from patients into the system; d) integration with modules for blood sampling for biobanking for genetic and proteomic research; and e) international collaborations: MINAP (United Kingdom), Infarctus Regiszter (Hungary), and ACTION (United States).
Registries From the Working Groups of the Spanish Society of CardiologyThe working groups of the Spanish Society of Cardiology have been conducting and publishing their activity registries annually. These registries have allowed us to know the trends in cardiovascular care in the last 2 decades and the regional differences with regard to the use of different cardiovascular interventions such as percutaneous coronary intervention, pacemakers, ablations, implantable cardioverter defibrillators, or heart transplant. However, these are not patient-level registries, in general no clinical outcomes are reported, and reliance on data is taken for granted with no quality controls or audits done. The Swedish registry should be inspirational for our country as a pursuable goal that will improve the quality of care and enhance research potential.89–93
FINAL REMARKWe have reviewed preclinical and clinical research as currently conducted, stressing the corresponding limitations, highlighting the barriers, and suggesting potential solutions. From animal studies to computational models, from traditional RCT to pragmatic registry-based RCT, from paper-based dedicated registries to EHR-linked national registries, all these developments contribute to the generation of evidence and ultimately to achieving a better quality of care for all (Figure 3).

It is absolutely clear for all that continuous innovation and change is needed in the way we conduct clinical research. A strong commitment to do this is demanded from all health professionals, investigators, politicians and society in general.
We call to action!
«Las ideas no duran mucho. Hay que hacer algo con ellas»
(“Ideas do not last long; something must be done with them”)
Santiago Ramón y Cajal
CONFLICTS OF INTERESTE.R. Edelman is funded in part by a grant from the National Institutes of Health (R01 GM 4903). The rest of the authors doesn’t have conflicts of interest to declare.