Keywords
INTRODUCTION
It has been known since the 1960s that myocardial infarction and, in general, all atherosclerotic disease, has an important hereditary component. Numerous studies in families and twins1-3 have described the risk of having a twin or a relative with coronary artery disease (Table 1).

Identification of the genes responsible for increased risk, however, has been a slow and difficult process, which has accelerated in the last decade thanks to advances in biotechnology, which have facilitated the detection of changes in the DNA sequence that can have a pathogenic effect.3 These changes, which we call mutations or polymorphisms, can be very subtle: sometimes a simple nucleotide (a simple amino acid in the encoded protein) among thousands is replaced (SNP or single nucleotide polymorphism); in others, the insertion or deletion of a segment takes place, or the repetition of tandem sequences (VNTR, variable number of tandem repeats). It can occur in the exon or encoding segment, the intron or promoter zone of the gene.
Mutation and polymorphism are, in fact, synonyms. Both are characterized by the coexistence of two varieties or alleles of the same gene, the natural or wild allele (wild type) and the mutant allele. But the term «mutation» usually is reserved for the changes that severely alter the function of the protein or enzyme encoded and one gene is enough to induce disease (monogenic or Mendelian disease). These diseases are rare and follow the laws of dominant or recessive Mendelian heredity. However, «polymorphism» designates common variations (which by definition occur in more than 1% of the population) with modest or minimum functional involvement, which cause an impairment (a genetic risk factor) when the organism must confront a greater metabolic effort or an environmental problem, such as a diet rich in cholesterol, stress, or smoking (environmental risk factor). Nevertheless, many polymorphisms do not have any functional consequences. The sum of several unfavorable polymorphisms can facilitate the appearance of a disease (in this polygenic), whose manifestation often requires the presence of a propitious environmental framework (multifactorial disease).
Arteriosclerosis is the most classic example (Figure 1) and a fair number of polymorphisms have been described that may be involved in their pathogenesis (Tables 2 and 3).4-6 The method used to describe an allele that confers risk is to identify a polymorphic gene as a possible candidate and demonstrate its statistical association with the clinical phenotype (coronary artery disease) and with the intermediate phenotype (biological values that depend on the enzyme).

Fig. 1. Etiology of coronary atherosclerosis: genetic and environmental factors. The polymorphic genes are shown in the oval forms in italics. The thin arrows indicate interactions between environmental and genetic factors. AGN indicates angiotensinogen gene; apo-E, apolipoprotein E; AT1, angiotensin receptor; ACE, angiotensin-converting enzyme; FGN, fibrinogen; HDL, high-density lipoprotein; AHT, arterial hypertension; IRS-1, insulin receptor substrate; LDL, low-density lipoprotein; Lp(a), lipoprotein (a); MTHR, Tetrahydrofolate reductase; NOS, nitric oxide synthase; NADH, NADH oxidase; PAI-1, plasminogen activator inhibitor.


GENES RELATED TO LOW-DENSITY LIPOPROTEINS
The list begins with the genetic variations that affect the plasma concentration of low-density lipoproteins, rich in cholesterol (LDL), whose deposition and harmful effect on the vascular wall constitutes the central phenomenon of atherogenesis4,5 (Figure 2).

Fig. 2. Genes of low-density lipoprotein metabolism (LDL). The genes involved in the etiology of coronary atherosclerosis are shown in italics in a box. Lower figure: scheme of a spherical particle of LDL, the specific agent of atheroma, with its molecule of filamentous apo-B surrounding it, to which a chain with kringle IV rings is added, apo(a), to form the Lp(a) complex. Arrow 1: pathway of LDL formation and transport to LDL-r receptors of hepatocytes or peripheral cells; arrows 2: transport of LDL, small LDL (type B), and Lp(a) to the vascular wall, where, after undergoing oxidation, they are captured by the SR-B1 receptors of macrophages. Apo-B, E, (a) indicates apolipoproteins B, E, and (a); IDL, intermediate-density lipoproteins; LDL, low-density lipoproteins; LDL-R, LDL receptors; SR-A, scavenger receptors; VLDL, very-low-density lipoproteins.
LDL receptor
The first gene that was related with myocardial infarction was the gene that encodes the LDL receptors (LDL-R).7 The mutations of this gene entirely impede receptor synthesis in hepatocytes («null mutation») or they deform it in such a way that they are incapable to binding circulating LDL for its elimination in bile. Consequently, accumulation these particles takes place, which characterizes familial hypercholesterolemia (FH). In its heterozygotic form, which is relatively frequent (1 out of every 500 persons), the cholesterol concentration increases up to 300-500 mg/dL, and is twice as high in homozygotic patients, which are exceptional (1/1 000 000). This high concentration facilitates the passive entrance of macromolecules through the junctions of the endothelial cells into the subendothelial space, where they suffer chemical modifications (peroxidation lipid, glycation) that convert them into pro-inflammatory, pro-cytotoxic, and atherogenic particles.4-7
Shortly after discovering the molecular defect of familial hypercholesterolemia, which earned them the Nobel prize in 1984, Goldstein and Brown described the first mutations responsible for causing disease.8 Today more than 230 are known (see http://www.ucl.ac.uk/fh). These mutations present a broad regional variability. In Spain, in the Mediterranean region, 55% of the mutations detected in heterozygotic patients were «null mutations». The rest had mutations of a simple nucleotide, the most frequent being 1146 G/A, 1301 C/G, and 829 G/A.9
The severity of the hypercholesterolemia and coronary artery disease also is highly variable: sometimes myocardial infarction appears at very young ages; in contrast, some heterozygotic patients reach the seventh or eighth decade of life without complications. This depends on: a) the type of mutation («null mutations» are the most serious);9,10>/SUP> b) the interaction with other genetic factors, the most important of which seems to be a favorable plasma high-density lipoprotein concentration (HDL) and, perhaps, the additional inheritance of a mutant heterozygotic lipoprotein lipase,9 or c) the importance of the environmental factors (dietary cholesterol and fat), as indicated by the difference in the prognosis of relatives who have migrated compared with that of the natives.4
A common variant is also known, Pvu II polymorphism in the intron 15 (which allows a cutoff to be established by the Pvu II enzyme and gives rise to fragments of different lengths in the electrophoretic study: RFLP polymorphism (restriction fragment length polymorphism). Little is known of this variant, but it apparently influences the LDL concentration and coronary risk.11
Apolipoprotein b (apo-B100)
Another, less severe, cause of familial hypercholesterolemia is the Arg3500Gln mutation (and, exceptionally, Arg3531Cys) of apolipoprotein B (apo-B100), the molecule that acts as a ligand for LDL receptors (familial defect of apo-B or FDB). The mutation impedes recognition by the LDL receptor and makes its plasma elimination difficult. Its prevalence is one case per 250-1000 persons.4,6,7
However, some common polymorphisms of the apo-B the gene are known whose association with hypercholesterolemia and coronary artery disease is very suggestive, but the contradictory results of studies make it questionable. These are Sp I/D polymorphisms (insertion/deletion in the signaling peptide of apo-B), EcoRI, and MspI (RFLP polymorphisms).12,13
Some rare mutations (0.3/1000) encode truncated forms of apo-B100 (apo-B55) and lead to low cholesterol levels (hypobetalipoproteinemia or HBLP), which probably has a protective effect against coronary artery disease.4,7
Apolipoprotein E (apo-E) polymorphisms
Apo-E is a protein that shares with apo-B the function of binding of lipoproteins rich in triglycerides (very-low-density [VLDL] and intermediate-density lipoproteins [IDL]). It occurs in three main versions: apo-E3, the natural isoform, is the most common, and two mutant forms, apo-E2, in which arginine has been replaced by cysteine in position 158, and apo-E4, in which cysteine is exchanged for arginine in position 112.
The polymorphic allele apo-E4 has less affinity than apo-E3 for apo-B/E receptors and is associated with a slight increase in cholesterol and plasma triglycerides. Its relation with coronary artery disease is well-founded.14-16 Carriers of this allele have a coronary risk that is 40% greater than that of the apo-E3 or apo-E2 alleles.15 The 4S study (Scandinavian Simvastatin Survival Study) has confirmed the lethal effect of the apo-E4 allele in patients who have suffered myocardial infarction. the mortality associated with this allele was 15.7% at 5.4 years of follow-up, compared with 9% in the rest of patients. In addition, the apo-E4 allele allowed the prediction of a good response to lipid-lowering treatment: the mortality of the patients with this allele decreased from 15.7% to 6%, whereas in the rest the reduction in mortality was appreciably smaller (from 6% to 5.1%).16 This data confirms that the response to treatment with statins depends on the apo-E genotype, as has been previously described.17 It has also been reported that the apo-E4 allele reduces the likelihood of reaching very advanced ages, because its prevalence in ninety year-old people (11.6%) is lower than in middle-aged individuals (22%).18 On the other hand, the apo-E4 allele is accompanied by a slight increase in the risk of suffering Alzheimer´s disease.4,7
In contrast, the apo-E2 allele seems to have a favorable effect. A recent comparative study of the effect of a diet rich in cholesterol on patients with different apo-E1, B, C-III, E, and R-LDL genotypes has demonstrated that individuals with the apo-E2 allele have the lowest LDL levels and do not respond to increased cholesterol in the diet, whereas the apo B mutations have the highest elevations, particularly if they coexist with the apo-E4 allele.19 The apo-E2 allele has also been related to: a) hypobetalipoproteinemia (LDL<70 mg/dL), whose prevalence in the Framingham Offspring Study was 2%. The other cause of hypobetalipoproteinemia, truncated apo-B, is much less frequent,13 and b) familial dysbetalipoproteinemia (hyperlipoproteinemia type III), a rare disease that affects patients with the apo-E2 allele, is produced by a complete deficiency in apo-E and is associated mainly with peripheral vascular disease.
Apo(a) polymorphism
The lipoproteins (a) Lp(a) are a small subgroup of LDL particles that contain, in addition to a molecule of apo-B100, a molecule of apo(a) that is extraordinarily similar to plasminogen, but does not mediate fibrinolysis. This molecule presents numerous repeated sequences (a maximum of 37) in the kringle IV ring («kringle» in memory of a Danish cookie), the same sequence that is repeated 5 times in plasminogen.4,6,20
The Lp(a) are particularly noxious, because: a) they are attracted by fibrin deposited on intimal lesions; b) they have an antifibrinolytic effect, because they compete with plasminogen and block its lysis-potentiating effect on the clot in acute coronary accidents, and c) they annul the plasmin-activating effect on the TGF-β factor, which has an important inhibiting effect on the proliferation and migration of smooth muscle cells.7,20
The plasma concentration varies individually from less than 1 to more than 100 mg/dL (10 mg/dL average) and maintains a narrow inverse relation with the number of apo(a) rings. The figure is, however, very constant in each individual and not very susceptible to dietetic or pharmacological changes, because the genetic factor is responsible for 90% of the variability.7,20
As numerous epidemiological studies have already confirmed, the presence of elevated concentrations of Lp(a) (>30 mg/dL) is associated with an increase in the incidence of myocardial infarction, restenosis, and cerebral vascular accidents.4,21,22 It has even been suggested that it can be the main cause of 25% of myocardial infarctions in the U.S.22 It also has an unfavorable prognostic meaning. In the patients with myocardial infarction in the Scandinavian 4S clinical trial, the mortality at 5.4 years of patients with an elevated concentration of Lp(a) was twice as high as in the rest. It is even more striking that if two unfavorable polymorphisms coincided, Lp(a) and the polymorphic apo-E4 allele, the mortality was three times as high, which is a good example of the amplifying effect of the interaction between two genes. On the other hand, the presence of apo(a) polymorphism predicted a more effective response to treatment with simvastatin, with a reduction in mortality of 50%. This reduction rose to 80% if the patient had both unfavorable polymorphisms, apo(a) and apo-E.16
GENES RELATED TO HIGH-DENSITY LIPOPROTEINS (HDL) AND INVERSE TRANSPORT
The Framingham study established that low plasma HDL concentration is one of the most powerful factors of coronary risk, as much or more than high LDL levels. It is attributed to the important protective role of these particles, which are responsible for the inverse transport of cholesterol from the vascular wall to hepatic receptors SR-B1 (to scavenger receptor type b1) for its elimination in bile.23 Plasma concentrations of HDL, and the effectiveness of inverse transport, depend on physical activity or alcohol consumption, but they have an important genetic component in 35%-50%, in which a fair number of polymorphisms are involved4,7 (Figure 3).

Fig. 3. Genes of the metabolism of HDL and triglycerides (TG). The polymorphic genes that encode the enzymes and receptors of TG metabolism are shown in italics and are involved in atherogenesis. Arrow 1: pathway of inverse cholesterol transport from the atheroma plaque to the liver, for elimination by bile. Their effectiveness depends on the production of the enzymes that facilitate the extraction of cholesterol from macrophages (ABC1) and its incorporation into HDL particles (LCA, HL) of the HDL components (apoproteins A1-CIII-A4, PONA), or specific hepatic receptors (SR-B1); arrow 2: pathway of alternative cholesterol transport from HDL to VLDL and IDL, regulated by the CETP enzyme. Box, transformation pathway of particles rich in triglycerides, VLDL, and IDL, in LDL, under the control of lipoprotein lipase (LPL). ABC1 indicates ABC1 transporter; Apo, apolipoprotein; CETP: cholesterol ester transfer protein; HL, heparin lipase; LCAT, lecithin acyl transferase enzyme; IDL, intermediate-density lipoproteins; LDL, low-density Lipoproteins; LDL-R, LDL receptors; SR-B1, scavenger receptors; VLDL, very-low-density lipoproteins.
Lipoprotein lipase and triglycerides
Lipoprotein lipase (LPL) is the main enzyme of the catabolism of lipoproteins rich in triglycerides (VLDL and IDL). Main twos have functions: a) it catalyzes the release of triglycerides (TG) by the VLDL when they pass through the capillaries and degrades them to remnants or IDL, and then to LDL, and b) once its enzymatic function is complete, it separates from its anchorage to the endothelium and as an IDL ligand for their uptake and elimination by hepatic receptors.4,7,23
The gene of the lipoprotein lipase is particularly prone to mutations. Close to 40 have been described (see: www.ncbi.nlm.nih.gov/omim). Some are located in the C-terminal segment (residues 313 to 448) and alter the ligand, and others are in the N dominion (from residue 1 to 312) and depress enzyme function and decrease VLDL and IDL catabolism, which causes accumulation of these particles in plasma (TG increase) and decrease of HDL.4,7
Three mutations located in the N dominion are known. Asp9Asn and Asn291Ser, whose prevalence in the heterozygotic form is 3%-5%, are associated with a 20%-30% increase in plasma TG, a reduction of the HDL-C to 10 mmol/L and an increase in the risk of ischemic heart disease (relative risk 1.3). Gly188Glu, the least frequent (1/1.000), depresses enzymatic activity more, inducing a greater increase in TG (80%), a greater decrease in HDL-C (25 mmol/L), and a more important increase in the coronary risk (relative risk 5.1).24
The mechanism of the relation between the increase in plasma TG (increased VLDL and IDL) and coronary artery disease continues to be debated. Several explanations have been proposed: a) it is suspected that the incorporation to the arterial wall of the smallest molecules of VLDL and IDL (VLDL residues), which also contain cholesterol although in smaller proportion, has an atherogenic effect comparable to that of LDL,25 but it is difficult to dissociate this effect from the important metabolic modifications that the TG induce (VLDL and IDL) in other lipoproteins, like HDL and LDL; b) the decrease in HDL, which has a close inverse relation with TG, attributable to the effect of TG (and insulin) on the catabolism of apo-A1, the apolipoprotein of the HDL; or to the greater activity of the CETP enzyme, which channels cholesterol transport to alternative pathways at the expense of inverse HDL transport (see below), or c) increased small, dense LDL (type B LDL), which may have major atherogenic potential due, possibly, to its greater capacity for penetrating the vascular wall and its easy oxidation.25-27
Multiple regression analyses seem to confirm that TG (VLDL and IDL) are a third factor of epidemiological risk, independent from the LDL and HDL, although the results are not very noteworthy. The effect is more evident when post-prandial TG are determined, which better reflect individual capacity for metabolic response.27 A recent meta-analysis of 17 studies summarizes the findings that support this concept.24
These polymorphisms of lipoprotein lipase have an interesting interaction with obesity, as indicated by the EARS study, because the TG elevation induced by excess weight is accentuated in carriers of the LPL-291S mutation.26
A fourth variant, the exonic polymorphism Ser447Ter, is located in the C-terminal end and affects ligand function. It has a special interest because, aside from being the most frequent (its prevalence in the general population is 20%), it is associated with a slightly higher HDL concentration (0.04 mmol/L), lower TG (8%), and a reduction in the risk of coronary artery disease (relative risk, 0.8). This protective effect is attributed to its greater affinity for receptors, which facilitates the elimination of VLDL particle residues.24 This variation is intimately linked to LPL/HindIII polymorphism (RFLP polymorphism).4,7
Cholesterol ester transfer protein (CETP) and alcohol
HDL have two essential enzymes for inverse transport: LCAT (lecithin-cholesterol acyl transferase), which facilitates cholesterol uptake by the vascular wall and initiates the inverse transport pathway, and CETP (cholesterol ester transfer protein), which promotes the transfer of cholesterol from HDL and LDL (rich in cholesterol) to VLDL and IDL (rich in TG), and initiates an alternative catabolic pathway for hepatic elimination that is less efficient than the inverse transport pathway.4,7
The known variations in LCAT do not seem to be more important, although a mutation, LCAT-Gly230 Arg, has been described that is responsible for 5% of cases of very low HDL.28 A TaqI-B polymorphism of intron 1 of the CETP gene (CETP/Taq1) is also known, which increases the activity of the enzyme and has a pro-atherogenic effect.29 This effect is attributed to: a) activation of CETP diverts cholesterol from inverse transport to the alternative pathway, with which the HDL decrease; b) it enriches the cholesterol of the particles that transport TG (VLDL and IDL), increasing their atherogenic potential , and c) deprives LDL of cholesterol, converting them into smaller, dense particles (B pattern LDL) that are especially atherogenic.4,7,26 It is suspected that post-prandial TG elevation and hypertriglyceridemia (which lasts 8 h so we are almost always in post-prandial period) have an important role in the genesis of atherosclerosis, partly because of its capacity to activate CETP.28
As the REGRESS study (Regression Growth Evaluation Statin Study) has demonstrated, carriers of the B1B1 genotype (35% frequency) have an elevated enzyme activity, high TG levels and low HDL levels, and present a greater progression of coronary atherosclerosis than carriers of B1B2 and B2B2. The presence of this allele also allows the prediction of a better response by angiographic lesions to treatment with statins.29
This polymorphism has a notable interaction with alcohol. As the ECTIM study (Etude Control-Temoigne l´Infarctus de Myocarde) has shown, the relation between CETP polymorphism and HDL are only manifested in persons who drink. However, it is not observed in non-drinkers. This indicates that the beneficial effect of moderate alcohol intake on HDL could be related to its depressive effect on CETP.30
Paraoxonase (HDL-PON1)
Paraoxonase/aryl esterase is a specific enzyme of the HDL (associated with the apo-A1) that is capable of hydrolyzing lipid peroxides and destroying the proinflammatory molecules produced by LDL oxidation, which is why it is suspected that it may have a relevant role in the etiopathogenesis of atherosclerosis.31 It is normally detected in the arterial wall and its concentration increases massively in atheroma, possibly in response to increased oxidative stress.31
The enzymatic activity of PON1 presents an interindividual variation of 10%-40%, partly attributable to the presence of two polymorphisms: PON1-192, with substitution of arginine by glutamine in codon 192 (A/G 192), that gives rise to an R isoenzyme with arginine and a mutant isoenzyme Q, and the PON1-55 that replaces leucine (L) by methionine (M) in codon 55. It has been confirmed by coincubation experiments that the capacity of HDL particles to protect against the oxidative modification of LDL is clearly superior in particles from QQ/MM homozygotes than from RR/LL homozygotes.32
Although some studies like ECTIM33 deny the association of polymorphism 192 with coronary artery disease, more recent ones indicate that the RR/LL genotype can be an important independent risk factor, particularly in patients with diabetes type 2, in which PON1 enzymatic activity and plasma levels are usually decreased.34 The REGICOR study has confirmed that the R allele increases the risk of infarction in diabetic patients.35 It is possible that the increased risk (which can be 60%) is only detected in smokers.36 This gene/environment interaction can be attributed to the deleterious effect of tobacco on paraoxonase, which annuls the effect of interindividual differences.
ABC1 transporter
Transporter ABC1 is a protein that intervenes decisively in cholesterol egress from cells, because it forms a channel that allows its passage outside the membrane, where it is transferred to the particles that arise from HDL after LCAT sterification. The confirmation that Tangier´s disease, a rare Mendelian disease that courses with very low HDL levels and premature coronary disease, is due to a mutation of the ABCA1 transporter gene propitiated the search for common polymorphisms of this gene that could influence coronary risk.4 The A -477T/C ABCA1 polymorphism was identified recently, whose TT and TC genotypes are associated with a modest reduction in HDL and apo-A1 levels, but show a very strong correlation with the severity of coronary artery disease, to judge from the number of angiographic lesions.37
Polymorphisms of apoproteins AI/CIII/AIV and hepatic lipase
Variants of the hepatic lipase and apo-A1/CIII/AIV loci (whose genes encodectural components of HDL) have been identified, which can influence the concentration of HDL and could modify the frequency of coronary artery disease.38 It should be remembered that there is a rare Mendelian disease, apo-AI deficiency, in which HDL is virtually absent (null allele) in the homozygotic form, which leads to severe premature coronary artery disease.4,7
ARTERIAL HYPERTENSION. RENIN-ANGIOTENSIN-ALDOSTERONE AND SYMPATHOADRENERGIC SYSTEMS
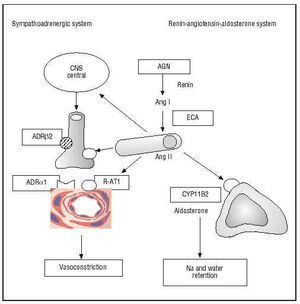
Arterial hypertension and ventricular hypertrophy were among the first coronary risk factors described in the Framingham study. It is thus logical that molecular variants of the genes of the renin-angiotensin-aldosterone and sympatho-adrenergic systems have undergone intense scrutiny (Figure 4).

Fig. 4. Genes of the sympatho-adrenergic and renin-angiotensin-aldosterone systems. In italics in the boxes are shown the polymorphic genes that encode enzymes or receptors involved in the pathogenesis of coronary artery disease. AGN indicates angiotensinogen; Ang, angiotensin; ADR, adrenergic receptor; CYP11B2, aldosterone synthase enzyme; ACE, angiotensin-converting enzyme; R-AT1, AT1 receptor of angiotensin II; CNS, central nervous system.
Angiotensinogen
Polymorphism M235T of the angiotensinogen (AGT) gene has a clearer relation with hypertension, although the effect seems to be weak. However, its relation with coronary artery disease is doubtful.39-41 It has been reported that genotype 235 TT doubles the risk of coronary heart disease and myocardial infarction,40 although the Copenhagen City Heart Study, with more than 2800 patients, has been negative.41
I/D polymorphism of the angiotensin-converting enzyme (ACE)
One of the best studied genetic variants is the insertion/deletion (I/D) polymorphism of the ACE gene, the ectoenzyme of endothelial cells that intervenes in the generation of angiotensin II and degradation of bradykinin, two vasoactive peptides with important effects on cellular proliferation and thrombosis. This polymorphism is characterized by the presence or absence of an Alu sequence of 287 base pairs in intron 16. It gives rise to three different genotypes: II/ID/DD. The carriers of allele D present an increase in plasma ACE activity and cardiac and tissular ACE (which represents 80% of ACE activity) and thus are exposed to higher angiotensin II levels. In the DD genotype, present in 28%-31% of the individuals (Table 2), the levels are twice as high as in the II genotype.4,42
Since 1992, when the association of the DD genotype with an increased risk of myocardial infarction was reported,42 numerous studies have been published on the role of allele D in the pathogenesis of coronary artery disease and myocardial infarction,43-48 postinfarction remodeling,49 postinfarction dispersion of QT and sudden death,48,50 restenosis after percutaneous intervention,51 insulin resistance and diabetes,52,53 arterial hypertension,54 or left ventricular hypertrophy.55 The ECTIM study, based on the MONICA registry, confirmed that the DD genotype is a powerful risk factor for myocardial infarction in patients under 65 years,42 above all in the absence of other risk factors, in which case the risk increases three-fold. It has been reported that the DD genotype can be the most important coronary risk factor in patients with type 2 diabetes.53 However, the ACE gene does not seem to appreciably influence arterial hypertension, which is why it is possible to attribute the atherogenic effect of angiotensin II to its metabolic action (mitogenic) on the vascular wall, more than to its systemic hypertensive effect.
On the contrary, some broad studies like the Physician´s Health Study (>3500 doctors),43 or the ISIS-3 study made in 11 000 subjects,46 deny that there is any relation between this polymorphism and myocardial infarction. Of two large meta-analyses that have been published, one concludes positively,44 and the other, which is more recent and includes more than 32 000 persons of white race,45 finds a significant increase in risk among the carriers of the DD homozygotic genotype. Nine years after the initial description,42 and although experimental and clinical tests provide convincing evidence in favor of the causal role of ACE in coronary artery disease, the implication of ACE polymorphism has not yet accepted.
Type I angiotensin II receptor
The A1166C polymorphism of the AT1R gene (of the type 1 angiotensin II receptor) is probably associated with an increase in the sensitivity of the AT1 receptor, through which most of the effects of angiotensin II are exerted. Allele C is more frequent in hypertensive patients, and its relation with susceptibility to ischemic heart disease is indicative.47,48,56,57 The association of AT1R-CC genotype with ACE DD (2% of our population) can have a synergic effect.47,48
Chymase (CMA)
Chymase is an enzyme of the vascular wall and heart that has the capacity to convert Ang I into II and big ET-1 into ET-1. The existence of this enzyme explains why the production of tissular Ang II can remain normal in spite of complete ACE inhibition. Nevertheless, it has not yet been possible to confirm the association of CMA A-1905G polymorphism and myocardial infarction.58,59
Aldosterone synthase (CYP11B2)
Aldosterone has important cardiovascular effects. Aside from its effect on blood pressure and volemia, its effect on fibrosis in hypertrophy and postinfarction is known.60 It is not strange, then, that the study of the C/T-344 polymorphism of the promoter region of the aldosterone synthase gene has awakened interest (CYP11B2/T-344C). This polymorphism has been implicated in the ventricular remodeling of arterial hypertension,61,62 or in postinfarction remodeling, which has not been confirmed.63 In a recent preliminary study of 16 genetic polymorphisms, regression analysis selected CYP11B2 polymorphism as one of the 9 polymorphisms related with angiographic coronary artery disease.64
Other genetic variants
Other polymorphisms in study are those of the gene of the β2 adrenergic receptor (ADRB2-Arg16Gly and Gln27Glu), of α-adducin (Gly460Trp), a protein of the internal layer of the plasma membrane that can be partly responsible for hypertension sensitive to salt; or of the endothelin (EDN1) and its A and B receptors (EDNRA and EDNRB). Their consideration as risk genes seems premature.5,65
GENES RELATED WITH INSULIN RESISTANCE(METABOLIC SYNDROME)
The metabolic syndrome, also called insulin resistance syndrome (or Reaven syndrome), is a particularly atherogenic syndrome that tries to explain the frequent association of four classic factors of coronary risk (the «death quartet» of Kaplan):66,67
1. Obesity that is predominantly visceral (waist/hip ratio>1.02 cm), related with intake and stress, which could be the necessary environmental factors for manifestation of the syndrome.
2. Hypertriglyceridemia, decrease of HDL, and increase of type B LDL (the so-called «lipid triad» or atherogenic dyslipemia). The increase in TG (by up to 60%) is attributed to an excess liver production of VLDL and IDL, stimulated by the greater availability of free fatty acids in adipose tissue. But it could also be attributed to a modification of lipoprotein lipase or CETP activity (see above).68-70
3. Insulin resistance, hyperinsulinemia, glucose intolerance, and type 2 diabetes. The greater availability of free fatty acids accentuates their consumption in skeletal muscle at the expense of glucose, whose increase in blood would stimulate secretion, and would make insulin resistance evident. Among the candidates genes for insulin resistance mutations of the insulin receptor have been proposed (more than 50 have been described), which would be rare monogenic causes; or the polymorphisms of FAB P2 (fatty acid binding protein), of lipoprotein lipase, of tumor necrosis factor alpha (TNF-α), or the DD genotype of ACE, but, above all, of the IRS gene (insulin receptor substrate).71 Most of the components of the syndrome are premonitory of type 2 diabetes, whose incidence reaches 15% versus 6.5% in the general population. Insulin resistance can precede type 2 diabetes by more than one decade.72
Hyperglycemia is atherogenic because it facilitates LDL glycation and converts highly damaging particles, and because it promotes the appearance of soluble advanced glycation end-products (AGE), which interact with specific endothelial receptors and increases the production of free oxygen radicals.73
4. Arterial hypertension is related with the increase in sympathetic tone produced by the central action of insulin, the activation of renin-angiotensin system, endothelial dysfunction, and ET-1 expression, or vasoconstrictive action induced by free fatty acids.
To this quartet can be added a dysfibrinolytic state imputable to the presence of high levels of plasminogen activator inhibitor (PAI-1), which are produced by endothelial and smooth muscle cells in response to the TG stimulus and insulin. This response is more intense in patients with PAI-1 polymorphism.68,70
Insulin receptor substrate (IRS-1)
The IRS-1 protein (insulin receptor substrate) acts as an intermediary between the insulin receptor and cascade of signals that determine membrane incorporation of the GLUT-4 protein (the transport channel in the absence of which the membrane is impenetrable to glucose). The cause of insulin resistance is with all probability lower IRS activity.72 The epidemiological studies already had found that insulin resistance and hyperinsulinemia notably increase the risk of myocardial infarction, even in non-diabetic patients,74 which is why when the G972R polymorphism of the IRS-1 gene was described, it was included immediately in the list of possible genetic risk factors. In this mutation a change of arginine for glycine in codon 972 gives place to two alleles, G and R. The prevalence in the general population of carriers of the mutation, that is to say, the GR genotype (RR is exceptional), is 6%-7%.71 Experimental and clinical studies have confirmed the association of this polymorphism with deterioration of IRS protein function, insulin resistance, and hyperinsulinemia, and with increased coronary risk. The mutation is found most frequently in patients with angiographic lesions (19% versus 6%-7%). It can be emphasized that the risk is multiplied by 7 if the patient is obese and by 27 if it also coexists with the clinical data of the metabolic syndrome.71
GENES RELATED WITH ENDOTHELIAL FUNCTION
The discovery that endothelial dysfunction can promote both the formation of atheroma plaques and the appearance of coronary accidents has been one of the most relevant contributions to the understanding of the genesis of atherosclerosis in recent years. The endothelium, aside from its macrovascular and microvascular vasodilator activity, also has an important antithrombotic, antiproliferative, and antiapoptotic action, and, in particular, an antioxidant function that can be decisive in protecting the vascular wall against the atherogenic effect of oxidized LDL and other harmful agents. All these functions are intimately related with the capacity of the endothelium to produce nitric oxide (NO), which counteracts the action of free oxygen radicals (superoxide anions, O2-) released during tissue metabolism.75 In normal conditions, NO production predominates over the generation of superoxide anions and maintains oxidative stress under control. If the production of NO is insufficient or the generation of free oxygen radicals is excessive, this balance can be broken, and the predominance of oxidative phenomena induces endothelial dysfunction with all its vasomotor and metabolic consequences (Figure 5).

Fig. 5. Genes that modulate endothelial function. The horizontal parallel arrows indicate the balance between the production of free oxygen radicals (O2-) by NADH oxidase and of nitric oxide (NO) by eNOS, which is under the control of angiotensin II and bradykinin (convergent arrows). The genes that encode enzymes are chown in italics in the boxes. The divergent arrows 1, 2 and 3 indicate the unfavorable effects of O2- on: a) smooth muscle cells (they potentiate the production of ET-1 and growth factors); b) LDL oxidation, and c) endothelial cells (which release proinflammatory cytokines and pro-thrombotic elements. In the four boxes in the lower part of the figure are shown the disturbances associated with endothelial dysfunction.
NADH/NADPH oxidase
The main source of production of free oxygen radicals is oxidative enzymes, notably NADH/NADPH oxidase, xanthine oxidase, myeloperoxidase, 15-lipo-oxygenase, or cyclo-oxygenase. A C242T polymorphism of the p22phox gene has been described, one of the elements of NADH microsomal oxidoreductase electron transport in smooth muscle cells. This reduces the production of superoxide anion in the vascular wall and improves the function of the coronary endothelium, which is why is suspected that it can reduce sensitivity to coronary artery disease.76,77
NO synthase
The capacity to counteract oxidative stress and maintain the integrity of endothelial function may depend on a polymorphism of endothelial NO synthase, in which a change in G → T takes place in the position that corresponds to nucleotide 894 (Glu 298 Asp), the NOS3 G894T polymorphism. In patients who require vasoconstrictive treatment during cardiac surgery, it has been confirmed that the vascular vasopressor response to phenylephrine (alpha adrenergic stimulation) is more intense in carriers of the 894 T (TT and GT) allele than in GG homozygotes, which indicates that vasodilator capacity is depressed due to the decreased NO production.78 Decreased NO3 enzyme activity could be a factor of coronary risk of the first magnitude,79 mainly in patients with vasospastic angina pectoris.80
Methylene tetrahydrofolate reductase (MTHFR), hyperhomocysteinemia, and folates
Hyperhomocysteinemia is now considered an important factor of coronary, cerebrovascular, and peripheral risk. The elevation of only 5 mmol/L (the normal upper limit is 15 µmol/L) increases coronary risk by 60% and 80% in men and women, respectively. This is a frequent disturbance (occurring in 5%-7% of the general population) that is attributed a decisive causal role in 10% of myocardial infarctions in the U.S. Its atherogenic effect is due to its accumulation in endothelial cells, where it increases oxidative stress and induces endothelial dysfunction.4,81
In half of the cases, the cause of hyperhomocysteinemia is the presence of the TT homozygotic genotype of the C677T polymorphism of MTHFR (5,10 methylene tetrahydrofolate reductase), which encodes a thermolabile variant of the enzyme MTHFR. This polymorphism is less efficient in its function of breaking down homocysteine into methionine for its elimination from cells. In these cases, plasma homocysteine increases by 25%, although this elevation apparently only occurs in the presence of a relative folate deficiency. The enzyme is dependent on folate and the mutated enzyme is less capable of using folates (thus increasing the dietary requirement); but homocysteinemia can be normal if folate intake is sufficient.81 The prevalence of this polymorphism is 10%-15% of the general population. Other mutations of enzymes that intervene in methionine metabolism, like cystathione synthase, are rarer.4
The second cause of hyperhomocysteinemia is the inadequate intake of vitamin cofactors like folic acid and vitamins B6 and B12. The environmental and genetic factors usually coincide.81 Other causes are hypothyroidism, kidney failure, the effect of certain medications like anticonvulsants, nicotinic acid, methotrexate, alcohol, and particularly tobacco, which interferes with vitamin B6 synthesis. This is a new atherogenic mechanism to add to the pernicious effects of smoking.
Several studies have reported that the TT homozygotic genotype increases coronary risk, especially if the level of folates is low;81 but contradictory results have been reported. A meta-analysis of 23 studies has been published with 6000 cases and 6000 controls that failed to confirm an association between the genotype and coronary artery disease.82 Nevertheless, a more recent Dutch meta-analysis, with almost 5000 cases, has reported a slightly positive result, and describes a gradual increase in coronary risk in the heterozygotic and homozygotic genotypes of allele T.83 It is possible that a definitive confirmation requires studies in large populations, or in vulnerable populations with folate deficiency.
Severe hypercysteinemias, with plasma concentrations of up to 100-400 µmol/L, capable of producing homocysteinuria, are exceptional Mendelian diseases, like homozygotic CBS deficiency, which, in addition to skeletal deformations and mental retardation, is associated with severe premature atherosclerosis and thromboembolic arterial and venous complications before the age of 30 years.4
GENES RELATED WITH THE INFLAMMATORY RESPONSE
Atherosclerosis is considered a chronic inflammatory process that appears as a reparative response to a metabolic, physical, or environmental vascular lesion. The severity and persistence of this local inflammatory response could be determined genetically, through genes that influence the capacity for producing inflammatory mediators (cytokines, adhesion molecules) or the capacity of monocytes and lymphocytes to respond to inflammatory stimuli, such as oxidized LDL, free O2- radicals, and endotoxins or lipopolysaccharides (LPS).
On the other hand, there is growing evidence that some systemic inflammatory or infectious processes can favor the appearance of coronary artery disease and its complications. This is the case of the low-grade chronic inflammatory response that is detected in clinical practice by increased levels of inflammatory mediators (C reactive protein [CRP], amyloidosis A, interleukin 6 [IL-6], fibrinogen [FGN], sICAM), although it is not clear if this inflammatory activity is the cause or effect of atheroma activity. An infection that has awakened interest is that caused by Chlamydia pneumoniae, one of the most frequent respiratory pathogens, whose association with coronary artery disease is confirmed by numerous seroepidemiological and experimental studies.
Among the polymorphisms that can influence local or systemic inflammatory response, the following are emphasized:84-87
Interleukin 6 (IL-6)
IL-6 occupies a central role in the genesis of the systemic inflammatory reaction and the acute-phase response, because it is the only cytokine that can stimulate the synthesis of all the other proteins that participate in the inflammatory response.84 A polymorphism has been discovered, G/C -174 of the gene of IL-6, that decreases the capacity for producing IL-6 and can cushion the inflammatory response. The baseline circulating levels of IL-6 in the CC genotype are 50% lower than in the GG genotype, and preliminary studies demonstrate that the risk of myocardial infarction is reduced by half (relative risk 0.54). It has been confirmed in vitro and in vivo that the variations in IL-6 production in response to LPS or IL-1 stimuli can originate important differences in the production of fibrinogen, endothelial adhesion molecules, or platelet aggregability.84
Interleukin 1 (IL-1β)
The polymorphisms of interleukin 1 (IL-1) could also influence the progression of atherosclerosis: the C/T -511 polymorphism of the gene of IL-1β and the polymorphism of the R-IL-1 receptor antagonist (aR-IL-1).85 It has been reported that patients seropositive to Chlamydia are more prone to suffering myocardial infarction if they carry the IL-1 C/C genotype and/or the 2 or 3-repeat allele of aR-IL-1. Apparently, the atherogenic effect of the infection is only manifested in the presence of this polymorphism, which confers the necessary susceptibility. This could explain the sometimes conflicting results of studies on the association between Chlamydia and coronary artery disease.85
CCR5 chemokyne receptors and CD14 monocytes
Another gene under study is that of the receptors of CCR5 chemokyne. The D-CCR5 polymorphism (deletion of 32 bp) can have a protective effect against myocardial infarction, which is attributable to the attenuation of the inflammatory response and slowing of atherosclerosis.86
A polymorphism, -260 C/T, has also been identified in the promoter of the CD14 receptor gene, which could modify the response of monocytes/macrophages to infectious stimuli and proinflammatory cytokines, and, consequently, increase their endothelial adhesiveness, capacity for infiltration and recruitment in the vascular wall, and transformation into macrophages. By flow cytometry it has been possible to confirm that the density of monocyte CD14 receptors is increased in TT homozygotes and that this genotype is most frequently found in patients who have suffered myocardial infarction.87
GENES RELATED WITH THROMBOSIS
Thrombosis and atherosclerosis are intimately related. The increase in procoagulant activity or the decrease in fibrinolytic capacity can contribute to the progression of the atheroma plaque and/or favor thrombus formation and coronary occlusion at the time of rupture.
Fibrinogen gene and tobacco
Fibrinogen has been found to be a powerful predictive factor of coronary artery disease, as much as classic risk factors can be. This reinforces the importance of its role in the progression of coronary stenosis and/ or the appearance of complications. It is involved in several mechanisms: a) it is the precursor of fibrin, which is deposited in the atheroma plaque and favors its growth through thrombus organization; b) it stimulates the proliferation of smooth muscle cells; c) it increases blood viscosity and has a rheological effect on endothelial function; d) it intervenes in adhesion phenomena, and e) in addition, can influence the severity of acute coronary accidents, because thrombi formed with high fibrinogen levels are more resistant to lysis than those formed with low levels.7
It has been repeatedly confirmed that fibrinogen concentration depends partly on its genotype.88-90 The molecule is formed by two identical subunits (dimer protein), each of which consists of three polypeptide chains (α, β, γ), encoded in tandem by three different genes of the long arm of chromosome 4. Several intimately associated polymorphisms in the gene of the beta chain have been described (FNG B), notably the -455 G/A polymorphism (the guanine/adenine polymorphism in the promoter region) and its marker, Hae III (RFLP polymorphism), and the Bcl I polymorphism.
In the ECTIM European study, the carriers of allele -455 A, found in 20%-25% of the general population, had a higher level of fibrinogen (351 mg/dl) that those of the GG genotype (338 mg/dl). The figure was 388 mg/dl in the AA genotype.88 In other studies, the differences range from 1% to 10%.3
One of the most notable characteristics of this polymorphism is its important interaction with tobacco, because increased fibrinogen concentration is observed only in smokers (or is more apparent in smokers). Smoking is the main environmental determinant of plasma fibrinogen, and smoking cessation reduces it by 10%.4 This effect is attributed to the fact that hepatic synthesis of fibrinogen forms part of the acute-phase response mediated by cytokines, because the gene has a promoter region sensitive to the IL-6 stimulus. Smoking promotes hepatic synthesis through its important proinflammatory stimulatory effect on IL-6 production.4,88 This effect could accentuate the coexistence of the -148 C/T FGN B polymorphism located in the promoter region sensitive to IL-6. The coexistence of these two polymorphisms explains why some smokers have such high fibrinogen concentrations.88
Although the relation between the genotype and fibrinogen (intermediate phenotype) has been confirmed repeatedly,88-90 the results of studies of the association with angiographic coronary artery disease or the incidence of myocardial infarction (clinical phenotype) have not been as convincing as could be expected. The majority are negative. In any case, its association with peripheral vascular disease has been described in the Edinburgh Artery Study,4,89 with myocardial infarction in the French branch of the ECTIM study,88 and with familial infarction in a selected population of GISSI-2.90
Polymorphisms of factors VII and XIII
Factor VII participates with tissue factor at the onset of the coagulation cascade (extrinsic pathway). Two polymorphisms can have an important protective role against myocardial infarction, because they are associated with a notable decrease in plasma levels of factor VII and propensity to thrombosis. This is the FVII R353Q polymorphism, with substitution of arginine (R) by glutamine (Q) in codon 353 of exon 8, and the 5´F7R polymorphism, which includes the insertion of 10 bp in position -323 of the 5´ region of the promoter, with two alleles, A1 (without the insertion) and A2 (with the insertion). Persons with the QQ or A2A2 homozygotic genotype have plasma levels of the VIIa factor (activated) that are 70% lower than in RR or A1A1 homozygotes. This important reduction apparently has a significant clinical effect, because the Q or A2 allele is associated with a reduction in the risk of myocardial infarction by half. The A2A2 genotype confers greater protection, which reduces the risk by 70% compared with genotype A1A1.91 This explains why some patients do not suffer myocardial infarction although they have severe coronary atherosclerosis.91,92 Other studies, however, deny any association.93
The contribution of the Val34Leu polymorphism of factor XIII is more debatable, although a protective effect against infarction and venous thrombosis has been described.94,95 This factor intervenes in the final phase of the coagulation by stabilizing the fibrin clot and confers more resistance to fibrinolysis.
It has been noted that polymorphisms of factor VIII could also be involved in coronary artery disease. These polymorphisms and factor IX (intrinsic pathway) intervene in the propagation of the clot, or von Willebrand factor (VWF), the most decisive molecule in the interaction between the platelets and vascular wall.4
PAI-1 gene and triglycerides
The increase in plasma PAI-1 s considered an important factor of thrombotic and atherogenic risk, since it buffers the natural fibrinolytic activity of t-PA. This protein is under the control of the 4G/5G polymorphism of the PAI-1 gene, which is characterized by the presence of five guanine nucleotides in the promoter zone instead of four. The 5G allele reduces transcriptional activity and the production of fibrinogen, so in principle it can be considered to be a favorable change. The carriers of the 4G4G homozygotic genotype have PAI-1 levels more than 10-50% higher than those of 5G/5G, and a reduced plasma fibrinolytic capacity. As can be understood from studies of association, the carriers of the 4G allele would be more prone to suffering coronary artery disease,96-98 although the conclusions of ECTIM97 have been negative. A recent meta-analysis demonstrates a weak propensity of the 4G allele to myocardial infarction.98
The existence of a very significant relation between plasma TG concentrations (and visceral obesity) and PAI-1 should be emphasized. Individuals with high TG levels and the 4G/4G genotype have higher PAI-1 levels than the carriers of the 5G/5G genotype (see metabolic syndrome). A sequence sensitive to TG has been identified in the promoter region of the gene that could explain this genetic/environmental interaction.
Gene of t-PA
The capacity of the endothelium to release large amounts of t-PA in acute coronary accidents to counteract massive fibrin deposits seems to be influenced by the Alu I polymorphism of the t-PA gene (insertion that contains a restriction site for the enzyme Alu I). In the Rotterdam study, its association with myocardial infarction has been observed, which is pending confirmation.99
Other genetic variants
The monogenic diseases produced by mutations of the hemostatic system increase the risk of hemorrhage or thrombosis (generally venous), but apparently do not have noteworthy effects on atherosclerosis. This is the case of hemorrhagic diseases caused by a VWF defect, the protein indispensable for formation of the platelet clot, and hemophilia A (factor VIII deficiency bound to chromosome X), which makes the formation of the fibrin thrombus difficult. Although it is been suspected that they have a protective effect, they are no guarantee against atherosclerosis. On the other hand, thrombophilic genetic variations, like Leyden factor V (present in 5% of the population) and the A20210G gene of prothrombin (in 2%), increase the risk of deep venous thrombosis but do not seem to influence the risk of arterial thrombosis or infarction.4-6
PLATELET POLYMORPHISMS
Among the genetic variations that can modify platelet activity, the polymorphisms of the three main glycoproteins (GP) of the platelet membrane must be noted: GP IIb/IIIa, the fibrinogen receptor; GP Ib-IX-V, the VWF receptor; and GP Ia-IIa, the collagen receptor.100-103
PlA1/PlA2 polymorphism of the gene of glycoprotein IIIa
Gene IIIa encodes one of the components of the receptor IIb/IIIa, the receptor that is bound to FGN and originates platelet aggregation and the platelet thrombus. The existence of a polymorphism of glycoprotein IIIa is known, in which the T/C substitution in position 1565 originates two variants, platelet antigens 1 and 2 (PlA1 and PlA2).
The Framingham Offspring Study has confirmed that the GP IIIa PlA2 polymorphism significantly increases platelet aggregability,101 which indicates that this allele, present in 16% of the population, can be an important genetic risk factor. Some studies have associated it with myocardial infarction, acute coronary syndromes, the risk of stent thrombosis, or ischemic complications in percutaneous interventions. Others have questioned the existence of any relation.101,102
Polymorphism of the GP Ib gene (Kozak polymorphism)
Receptor GP Ib-IX-V plays a critical role in the adhesion of platelets to the injured vascular wall in the initial phase of hemostasis, because it binds to the subendothelial matrix rich in VFW. The -5 T/C polymorphism in the Kozak sequence of GP Iba (the subunit of GP Ib-IX-V complex that binds to VFW) has been associated with an increase in the density of GP Ib-IX-V receptors in the platelet membrane, which could be a risk factor for platelet thrombosis in acute coronary syndromes or complications post-transluminal percutaneous coronary angioplasty. The carriers of allele -5C (carriers of the Kozak polymorphism) present unstable angina more frequently than those of the natural allele (5T). Determination of the genotype could facilitate risk stratification and the design of more suitable antiplatelet treatment.103
Polymorphism of the gene of glycoprotein Ia
GP Ia-IIa is the collagen receptor that stabilizes platelets adhered to the vascular wall. Some polymorphisms of the GP Ia gene increase by up to 10-fold the density of collagen receptors on the surface of platelets. Studies of the predisposition to acute coronary syndromes have obtained different results.100
CLINICAL IMPLICATIONS
The idea that individual genetic variability can play a role as a determinant of coronary risk is very attractive, above all in view of the fact that classical risk factors cannot account for more than 30%-50% of the cases of coronary artery disease.4 Numerous alleles of risk have already been described and the list of candidates genes continues to expand. Our knowledge, nevertheless, has important limitations. In most cases, the association with coronary risk or the intermediate phenotype is very modest or questioned. In spite of these limitations, a hypothesis is taking shape that the risk of suffering coronary artery disease depends on the number of unfavorable polymorphisms that an individual has. The possible consequences of this concept are important:
1. Analysis of the genomic profile (molecular diagnosis) can facilitate the diagnosis of individual genetic susceptibility from the sum of the risk alleles present. An example of what could be in the near future is illustrated by a recent preliminary study in which the risk is analyzed based on the 8 most relevant polymorphisms identified by the regression study, which are related to lipids (LPL Pvu and Hind III, CETP), the renin-angiotensin-aldosterone system (AGT, CMA, and aldosterone synthase) and thrombosis (PAI-1).62 Perhaps the definitive list of genetic factors must wait until the Human Genome Project-2 (1998-2003) offers a complete catalog of polymorphisms and we have adequate technology (microchips) for studying associations in thousands of individuals.
2. The description of the genetic bases (pathophysiology) can solve the problem of the etiological classification of coronary atherosclerosis, an old aspiration.
3. Genetic tests can be useful in the prediction of the effectiveness of a drug in a given patient (pharmacogenomics). The genotype is a factor to consider in interpretations of the results of clinical trials.
4. A different topic is to consider the possible benefit of early stratification of the genetic risk by means of DNA tests (systematic screening). The extent to which it can replace or complete traditional biochemical diagnosis is still a topic of debate.
5. The most ambitious goal of preventive medicine, genetic modifications designed to prevent infarction (gene therapy), is distant, but perhaps less than we think.
Correspondencia:
Prof. F. Navarro López.
Servicio de Cardiología (ICMCV). Hospital Clínico.
Villarroel, 179. 08036 Barcelona. España
E-mail: navarro@medicina.ub.es