TPM1 is one of the main hypertrophic cardiomyopathy (HCM) genes. Clinical information on carriers is relatively scarce, limiting the interpretation of genetic findings in individual patients. Our aim was to establish genotype-phenotype correlations of the TPM1 p.Arg21Leu variant in a serie of pedigrees.
MethodsTPM1 was evaluated by next-generation sequencing in 10 561 unrelated probands with inherited heart diseases. Familial genetic screening was performed by the Sanger method. We analyzed TPM1 p.Arg21Leu pedigrees for cosegregation, clinical characteristics, and outcomes. We also estimated the geographical distribution of the carrier families in Portugal and Spain.
ResultsThe TPM1 p.Arg21Leu variant was identified in 25/4099 (0.61%) HCM-cases, and was absent in 6462 control individuals with other inherited cardiac phenotypes (P<.0001). In total, 83 carriers (31 probands) were identified. The combined LOD score for familial cosegregation was 3.95. The cumulative probability of diagnosis in carriers was 50% at the age of 50 years for males, and was 25% in female carriers. At the age of 70 years, 17% of males and 46% of female carriers were unaffected. Mean maximal left ventricular wall thickness was 21.4 ±7.65mm. Calculated HCM sudden death risk was low in 34 carriers (77.5%), intermediated in 8 (18%), and high in only 2 (4.5%). Survival free of cardiovascular death or heart transplant was 87.5% at 50 years. Six percent of carriers were homozygous and 18% had an additional variant. Family origin was concentrated in Galicia, Extremadura, and northern Portugal, suggesting a founder effect.
ConclusionsTPM1 p.Arg21Leu is a pathogenic HCM variant associated with late-onset/incomplete penetrance and a generally favorable prognosis.
Keywords
Hypertrophic cardiomyopathy (HCM) is a common genetic disorder (prevalence >1/500), with wide phenotypic and locus heterogeneity.1–3 The TPM1 gene (encoding α-tropomyosin) is considered one of the main causative HCM genes; nevertheless, it is a relatively rare etiology, accounting for 1% to 5% of the cases with this phenotype.3
Available clinical information on most of the patients carrying TPM1 variants is restricted to a single pedigree or a few index cases for each variant,4–13 which limits assertive clinical interpretation of the genetic findings. The exception to this rule is the TPM1 p.Asp175Asn variant identified in several Finnish HCM patients because it is the single founder effect described in this gene to date.14,15 Genotype-phenotype correlation studies of founder variants in HCM populations have contributed to a better understanding of the clinical course and prognosis associated with this variant.16–22
At our center, we identified the variant TPM1 p.Arg21Leu by next-generation sequencing in several individuals with HCM, including homozygous carriers. Although we undertook genetic studies from several countries around the world, the p.Arg21Leu variant cases predominantly came from hospitals in northwest Spain and Portugal, raising the hypothesis that it could be a founder effect. To date, this variant has been classified in public databases as of uncertain clinical significance (5 independent submitters) and likely pathogenic (only 1 submitter),23 while the associated-phenotype is still unknown. Our main objective in this study was to describe the phenotypic features and associated prognosis of the TPM1 p.Arg21Leu variant in a series of pedigrees.
MethodsThis is a descriptive study of a series of families carrying the variant TPM1 NP_001018005.1: p.Arg21Leu (c.62G >T). This study was performed in accordance with the principles of the Helsinki Declaration, and is part of the research line registered with the number 2012/139 in the Research Ethics Committee of Galicia, Spain. Informed consent was obtained for all participants.
Study population and bioinformatic analysisFrom March 2008 to September 2018, the TPM1 gene was sequenced in 10 561 consecutive index cases with different inherited heart diseases from distinct hospitals around the world referred to our center for molecular diagnosis.
All exons and intronic boundary regions of 213 genes () related to cardiomyopathies and sudden cardiac death (SCD) were studied by next-generation sequencing in the index cases. The minimum read-depth obtained was >30 (average from 250 x to 400 x) in deoxyribonuclei acid (DNA). Fragments that did not fulfill these criteria were sequenced by Sanger. Familial cascade genetic screening in relatives was performed using Sanger sequencing. A multidisciplinary team performed bioinformatics analysis and clinical interpretation. Information regarding allelic frequency in the general population (controls) was considered based on Genome Aggregation Database (gnomAD) and TOPMed Program populations. The pathogenicity classification of the variants was in accordance with current recommendations, which consider the prevalence of the variant in affected vs control individuals, familial cosegregation data, functional studies, and in silico predictors, among other criteria.24 Pathogenic/likely pathogenic variants in any of the sequenced genes were described, as well as those of unknown clinical significance in 18 priority HCM genes ().
Additionally, contact was made during the first Iberian meeting of cardiomyopathies (Óbidos, Portugal, March, 2017) to collect information from TPM1 p.Arg21Leu carriers identified at other Portuguese and Spanish centers.
Phenotype characterizationClinical data from index cases and family members were reviewed, and their pedigrees were constructed. All carrier families were included, regardless of the length of follow-up or the number of family members evaluated. We analyzed the factors related to penetrance and disease expression in HCM: age, sex, genotype, clinical features, and SCD risk factors. Early onset was defined as diagnosis in persons younger than 35 years.25 The diagnostic criteria and SCD risk stratification model for HCM followed the recommendations of the European Society of Cardiology (ESC).3 All probands fulfilled conventional diagnostic criteria for HCM with ≥ 1.5cm wall thickness on echocardiography in at least 1 myocardial segment. Carrier relatives with left ventricular wall thickness ≥ 1.3cm were considered to be diagnosed with HCM. Relatives with only minor electrocardiogram (ECG) changes or normal ECG and normal echocardiography, regardless of their carrier status, were considered clinically unaffected. Relatives who had not undergone diagnostic evaluation were classified as nonclinically evaluated. SCD risk score (ESC calculator) was obtained for carriers with an HCM diagnosis and available clinical data. Other SCD risks were also considered2,26: left ventricular apical aneurysm, end-stage HCM, and extensive late gadolinium enhancement (LGE) (defined here as ≥ 3 affected cardiac segments) on magnetic resonance imaging.
The cumulative probability of occurrence of cardiovascular death or equivalent (SCD, appropriate defibrillator shock, heart failure death, stroke-related death, and unspecified cardiac death) and heart transplant was estimated with the Kaplan-Meier method, and factors were compared using the log-rank (Mantel-Cox) method. Survival was calculated from birth. A 2-sided P value<.05 was considered to indicate statistical significance. TPM1 p.Arg21Leu carriers (index cases and relatives) and all clinically affected first- and second-degree relatives without genetic testing were included for this evaluation. The cumulative probability of an HCM diagnosis in carriers by age was estimated also by the Kaplan-Meier method. A 2-point logarithm of the odds (LOD scores) was calculated in all families using the PARAMLINK package for R software. The model was set with θ=0, phenocopy rate=0.005 and 2 different penetrance values: 0.80 and 0.95. An indeterminate status was assigned to family members who were reported only with diagnostic suspicion, as well as to males younger than the age of 50 years and females younger than age 55 years who did not fulfill the clinical criteria for HCM and could subsequently develop the disease.
Information about the region of origin of each index case was also collected to estimate the number of carrier families in different regions and the geographic distribution of the variant in Portugal and Spain.
Statistical analyses were performed with the software R version 3.4.3 (R Foundation for Statistical Computing, Vienna, Austria) and Health in Code (HiC) mutations version 7.7.6 (Health in Code S.A., A Coruña, Spain).
RESULTSTPM1 p.Arg21Leu was identified in 25/10 561 (0.23%) consecutive index cases with different inherited heart diseases sequenced in our center. The variant was present in 25/4099 (0.61%) HCM-probands, and absent in 6462 index cases with other cardiac phenotypes (0/3830 dilated, left ventricular noncompaction, and arrhythmogenic cardiomyopathies; 0/1590 channelopathies, and 0/1042 other inherited heart diseases) (P<.0001). This variant appeared in simple heterozygosis in 10/62 784 (0.016%) individuals from the TOPMed program population, and in 5/120 158 (0.004%) individuals (age range 55-65 years) from the gnomAD population (non-TOPMed samples).
Further, we included 6 additional index cases identified in other collaborating hospitals. In total, 83 carriers (50.6% male) were identified—31 index cases. Detailed clinical data were described for 67 carriers; 44 (65.7%) were clinically affected ( and ).
Twenty-eight pedigrees were constructed (familial data were not reported in 3 probands), and familial cosegregation of the variant was documented with an autosomal dominant inheritance pattern (combined LOD score=3.95). All pedigrees, study population, specific LOD scores, individual clinical data, and SCD risk scores are available in the .
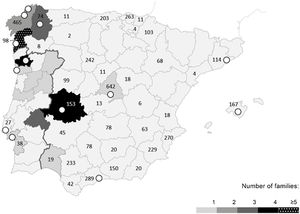
Index cases were evaluated from 13 hospitals (4 Portuguese and 9 Spanish). The reported origin of the families was concentrated in the western part of the Iberian Peninsula (the region of Braga in northern Portugal, as well as the Spanish regions of Galicia and Extremadura) (figure 1). We did not identify the TPM1 p.Arg21Leu variant in samples referred from hospitals of other parts of the world.
 of the families carrying the TPM1 p.Arg21Leu variant. Portugal and Spain maps divided by region. Concentration of families (origin) is represented in gray scale. White circles indicate the reference hospitals where the index cases were identified. Numbers (n) represent the hypertrophic cardiomyopathy studies requested by each region.")
Geographic distribution (by origin) of the families carrying the TPM1 p.Arg21Leu variant. Portugal and Spain maps divided by region. Concentration of families (origin) is represented in gray scale. White circles indicate the reference hospitals where the index cases were identified. Numbers (n) represent the hypertrophic cardiomyopathy studies requested by each region.
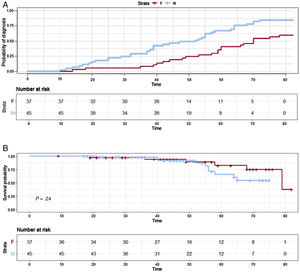
The cumulative percentage of diagnosis in carriers by age and sex is shown in figure 2. At the age of 30 years, 25% of male and 6% of female individuals had been clinically diagnosed with HCM. The percentage rose to 50% at the age of 50 years for male vs 25% for female carriers. At the age of 70 years, approximately 17% of males and 46% of females were still unaffected. The mean age at diagnosis was 47.3±18.8 [range 11-73] years. Individuals diagnosed under the age of 35 years were 18% (12/67) of the carriers with detailed data.
Phenotypic features and survival function (below), both by age and sex. Male individuals are in blue and female in red. F, female; M, male.")
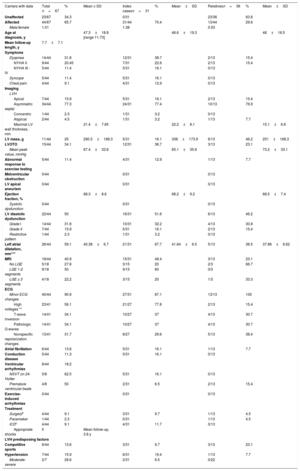
Clinical data obtained from 67 carriers (31 index cases and 36 relatives) are summarized in table 1. Forty-four carriers (44/67, 66%; 31 index cases and 13 relatives) met the diagnostic criteria for HCM by left ventricular hypertrophy (LVH) criteria, and 23 carriers (23/67; 34%) were unaffected.
TPM1 p.Arg21Leu clinical features (n=67)
| Carriers with data | Total n=67 | % | Mean ± SD | Index casesn=31 | % | Mean±SD | Relativesn=36 | % | Mean±SD |
|---|---|---|---|---|---|---|---|---|---|
| Unaffected | 23/67 | 34.3 | 0/31 | 23/36 | 63.8 | ||||
| Affected | 44/67 | 65.7 | 31/44 | 70.4 | 13/44 | 29.6 | |||
| Male:female | 1.01 | 1.38 | 0.53 | ||||||
| Age at diagnosis, y | 47.3±18.8 [range 11-73] | 46.6±19.3 | 48±16.5 | ||||||
| Mean follow-up length, y | 7.7±7.1 | ||||||||
| Symptoms | |||||||||
| Dyspnea | 14/44 | 31.8 | 12/31 | 38.7 | 2/13 | 15.4 | |||
| NYHA II | 9/44 | 20.45 | 7/31 | 22.6 | 2/13 | 15.4 | |||
| NYHA III - IV | 5/44 | 11.4 | 5/31 | 16.1 | 0/13 | ||||
| Syncope | 5/44 | 11.4 | 5/31 | 16.1 | 0/13 | ||||
| Chest pain | 4/44 | 9.1 | 4/31 | 12.9 | 0/13 | ||||
| Imaging | |||||||||
| LVH | |||||||||
| Apical | 7/44 | 15.9 | 5/31 | 16.1 | 2/13 | 15.4 | |||
| Asymmetric septal | 34/44 | 77.3 | 24/31 | 77.4 | 10/13 | 76.9 | |||
| Concentric | 1/44 | 2.3 | 1/31 | 3.2 | 0/13 | ||||
| Atypical | 2/44 | 4.5 | 1/31 | 3.2 | 1/13 | 7.7 | |||
| Maximal LV wall thickness, mm | 21.4±7.65 | 22.2±8.1 | 15.1±6.6 | ||||||
| LV mass, g | 11/44 | 25 | 290.5±168.3 | 5/31 | 16.1 | 338±173.9 | 6/13 | 46.2 | 251±168.3 |
| LVOTO | 15/44 | 34.1 | 12/31 | 38.7 | 3/13 | 23.1 | |||
| Mean peak value, mmHg | 67.4±33.8 | 65.1±35.6 | 73.2±33.1 | ||||||
| Abnormal response to exercise testing | 5/44 | 11.4 | 4/31 | 12.9 | 1/13 | 7.7 | |||
| Midventricular obstruction | 0/44 | 0/31 | 0/13 | ||||||
| LV apical aneurism | 0/44 | 0/31 | 0/13 | ||||||
| Ejection fraction, % | 68.3±8.6 | 68.2±9.2 | 68.5±7.4 | ||||||
| Systolic dysfunction | 0/44 | 0/31 | 0/13 | ||||||
| LV diastolic dysfunction | 22/44 | 50 | 16/31 | 51.6 | 6/13 | 46.2 | |||
| Grade I | 14/44 | 31.8 | 10/31 | 32.2 | 4/13 | 30.8 | |||
| Grade II | 7/44 | 15.9 | 5/31 | 16.1 | 2/13 | 15.4 | |||
| Restrictive pattern | 1/44 | 2.3 | 1/31 | 3.2 | 0/13 | ||||
| Left atrial dilatation, mm**a | 26/44 | 59.1 | 40.38±6.7 | 21/31 | 67.7 | 41.64±6.5 | 5/13 | 38.5 | 37.86±6.62 |
| MRI | 18/44 | 40.9 | 15/31 | 48.4 | 3/13 | 23.1 | |||
| No LGE | 5/18 | 27.8 | 3/15 | 20 | 2/3 | 66.7 | |||
| LGE 1-2 segments | 9/18 | 50 | 9/15 | 60 | 0/3 | ||||
| LGE ≥ 3 segments | 4/18 | 22.2 | 3/15 | 20 | 1/3 | 33.3 | |||
| ECG | |||||||||
| Minor ECG changes | 40/44 | 90.9 | 27/31 | 87.1 | 13/13 | 100 | |||
| High voltages ** | 23/41 | 56.1 | 21/27 | 77.8 | 2/13 | 15.4 | |||
| T-wave inversion | 14/41 | 34.1 | 10/27 | 37 | 4/13 | 30.7 | |||
| Pathologic Q waves | 14/41 | 34.1 | 10/27 | 37 | 4/13 | 30.7 | |||
| Nonspecific repolarization changes | 13/41 | 31.7 | 8/27 | 29.6 | 5/13 | 38.4 | |||
| Atrial fibrillation | 6/44 | 13.6 | 5/31 | 16.1 | 1/13 | 7.7 | |||
| Conduction disease | 5/44 | 11.3 | 5/31 | 16.1 | 0/13 | ||||
| Ventricular arrhythmias | 8/44 | 18.2 | |||||||
| NSVT on 24-Holter | 5/8 | 62.5 | 5/31 | 16.1 | 0/13 | ||||
| Premature ventricular beats | 4/8 | 50 | 2/31 | 6.5 | 2/13 | 15.4 | |||
| Exercise-induced arrhythmias | 0/44 | 0/31 | 0/13 | ||||||
| Treatment | |||||||||
| Surgeryb | 4/44 | 9.1 | 3/31 | 9.7 | 1/13 | 4.5 | |||
| Pacemaker | 1/44 | 2.3 | 0/31 | 1/13 | 4.5 | ||||
| ICDc | 4/44 | 9.1 | 4/31 | 11.7 | 0/13 | ||||
| Appropriate shocks | 0 | Mean follow-up, 3.8 y | |||||||
| LVH predisposing factors | |||||||||
| Competitive sports | 6/44 | 13.6 | 3/31 | 9.7 | 3/13 | 23.1 | |||
| Hypertension | 7/44 | 15.9 | 6/31 | 19.4 | 1/13 | 7.7 | |||
| Moderate-severe | 2/7 | 28.6 | 2/31 | 6.5 | 0/22 | ||||
ECG, electrocardiogram; ICD, implanted cardiodefibrillator; LGE, late gadolinium enhancement; LV, left ventricle; LVH, left ventricular hypertrophy; LVOTO, left ventricular outflow tract obstruction; MRI, cardiac magnetic resonance; NSVT, nonsustained ventricular tachycardia; NYHA, New York Heart Association; SD, standard deviation.
P value Fisher exact test for the subpopulations index cases and relatives, only shown [**] when P value <.05 comparing index cases and relatives.
The predominant left ventricular morphological pattern was asymmetric septal (34/44; 77%) and 16% (7/44) had apical hypertrophy. shows the distribution of maximum left ventricular wall thickness (in mm) by sex, genotype, and age at the last follow-up. Carriers were concentrated between 15 to 25mm of LVH and at advanced ages. Mean maximum left ventricular wall thickness was 21.4±7.65mm. Among the carriers diagnosed at <35 years, mean maximum left ventricular wall thickness was 27.04±11.2mm. Left ventricular outflow tract obstruction was reported in 34% of cases (15/44) and abnormal blood pressure response to exercise testing in 11% (5/44). Sixteen percent (16%; 7/44) had a pseudonormalization pattern of the mitral valve inflow, and only 1 case had a restrictive pattern. No left ventricular midventricular obstruction was observed.
ECG abnormalities were reported in 91% of the affected carriers; 13.6% (6/44) of them had atrial fibrillation, and 5 carriers (11.3%; 5/44) were reported to have conduction system disorders, 5 cases of first-degree atrioventricular block. One carrier with complete atrioventricular block after mitral valve surgery was not included in this subgroup because his atrioventricular block was considered secondary to surgery. He underwent pacemaker implant. Syncope was reported in 11.5% (5/44) of the affected carriers. Eight (8/44; 18%) of the affected carriers had ventricular arrhythmias; 5 of them with nonsustained ventricular tachycardia recorded on 24-hour Holter monitoring. There were no arrhythmias induced by the exercise stress test. There were no reports of appropriate shocks in a mean follow-up of 3.8 years among the 4 male patients who had implanted cardioverter-defibrillators for primary prevention; 3 of them had been diagnosed in adolescence and received the devices approximately 10 years after diagnosis. Two carriers with a cardioverter-defibrillator had high SCD risk (9.94%, and 6.37% over 5 years) and the other 2 had intermediate risk scores (both with 4.59% over 5 years), without additional SCD risk markers. SCD risk scores were obtained at the time of clinical decision-making.
Four carriers with early onset HCM (33%; 4/12) were competitive athletes (all football players) vs 2/32 (6.2%) carriers with late onset (P=.074) (, clinical features of the TPM1 p.Arg21Leu carriers diagnosed under the age of 35 years).
Only 2 clinical features showed significant statistical differences between index cases and relatives (table 1). Index cases had higher ECG voltages and more atrial dilatation than relatives.
Risk stratification and cardiovascular eventsSurvival analysis (figure 2) showed that less than 5% of our population had a cardiovascular death or heart transplant at the age of 30 years, this figure being 10% at 50 years for both sexes. Twenty-five percent of female individuals and 44% of males had had a major cardiovascular event at 70 years. No statistical difference between sexes was observed (P=.24).
Three cardiovascular deaths (1 SCD, 1 heart failure death, and 1 unspecified cardiac death) and 1 heart transplant (in total, 4/83; 5%) were reported in TPM1 p.Arg21Leu carriers. During a mean follow-up time of 7.7±7.1 years, no SCD or heart transplant was additionally reported. Twelve other cardiovascular events were reported in first- or second-degree relatives without genetic testing; 5 SCD, 2 heart failure deaths, 2 stroke-related deaths, and 3 unspecified cardiac deaths. Five carriers were reported to have had nonmajor cardiovascular events that were not included in the survival curves; 2 myectomies, 1 mitral valve replacement for left ventricular outflow tract obstruction due to systolic anterior motion, and 2 nonlethal strokes. Each event, age at occurrence and patient sex are specified in .
ESC SCD risk was calculated for all affected carriers (n=44). Most of them (77.3%; 34/44) had a low risk score (<4% risk over 5 years), 18.2% (8/44) had intermediate values (4-6% risk over 5 years), and only 4.5% (2/44) had high risk (>6% risk over 5 years). shows that carriers were concentrated at ranges with low SCD risk and advanced ages. Eighteen affected carriers (18/44; 41%) had a magnetic resonance imaging scan; 5/18 (27.8%) were reported without LGE, 9/18 (50%) had LGE in 1 or 2 cardiac segments, and 4/17 (22.2%) had LGE in ≥ 3 segments. No left ventricular apical aneurysm or end-stage HCM was observed.
Homozygous carriers and additional genetic variantsFifteen individuals (15/67; 17.9%) from 10 pedigrees had complex genotypes. Four had TPM1 p.Arg21Leu in homozygosity (4/67; 6%), and 12 had an additional genetic variant (12/67; 17.9%); 1 of the homozygous carriers also had the pathogenic variant MYBPC3 p.Asp75Asn. Clinical features of the homozygous carriers are described in .
Five genetic variants in the MYH7 gene were identified (p.Gly741Arg and p.Thr1019Asn, respectively classified as pathogenic/likely pathogenic variants; p.Lys351Asn, p.Tyr582Cys and p.Leu1333Val as variants of unkown clinical significance). Other pathogenic sarcomeric variants were identified: TPM1 p.Met281Thr, TNNT2 p.Arg278Cys, and MYL3 p.Met173Val. Each of these variants was identified in different pedigrees. All these variants have been reported with very low allelic frequency (<0.01%) in the general population, except MYH7 p.Lys351Asn. See complementary description of each variant in , showing clinical features of carriers with an additional genetic variant.
No cardiovascular death or heart transplant was reported in confirmed carriers with an additional genetic variant. Two SCD in first-degree relatives without genetic testing were reported in pedigrees with an additional pathogenic genetic variant (MYH7 p.Gly741Arg, and TPM1 p.Met281Val, respectively).
DiscussionHere we present the largest HCM population carrying the same TPM1 variant described in the literature to date. This study shows that the TPM1 p.Arg21Leu variant was significantly overrepresented in our HCM cohort compared with control populations, and familial cosegregation of the variant with the disease was documented with a significant LOD score.27 Taking into account these results, there are sufficient criteria to now classify p.Arg21Leu as clearly pathogenic ().
TPM1 p.Arg21Leu was identified exclusively in patients with the HCM phenotype. Most of the individuals in our population were simple heterozygous carriers with late onset HCM, mild to moderate phenotype, asymptomatic clinical course, and a low number of cardiovascular deaths and heart transplants. The annual incidence of cardiovascular events was 0.25% in the survival analysis, which suggests a better prognosis for TPM1 p.Arg21Leu than expected for the overall HCM population (≅ 0.5%/y).28,29 SCD risk scores were predominantly in the low risk range, followed by approximately 20% of the cases with intermediate scores. Only 2 carriers had high SCD risk.
In comparison, the Finnish TPM1 founder variant, p.Asp175Asn, was described in the literature as being associated with mild-moderate HCM phenotype and favorable prognosis; nonetheless, a higher penetrance in adulthood (91%-95%) was reported, based on 2 unrelated smaller cohorts.13,14 Other variants in the MYBPC3 and MYH7 genes previously reported as founder effects in HCM populations have shown a worse prognosis than TPM1 p.Arg21Leu.17–21 Mean age at diagnosis in individuals carrying some of these variants was also lower, approximately 1 decade less.
TPM1 p.Arg21Leu carriers with early onset HCM were predominantly male. No additional pathogenic genetic variant (except 1 who had a MYH7 variant of unknown clinical significance) was reported in this group. Their SCD risk scores were higher than the overall average, and they had more prominent LVH (both myectomies described in our study were in individuals from this group), but no cardiovascular death or heart transplant was documented among the carriers diagnosed under the age of 35 years. Other groups have described similar marked intrafamilial clinical heterogeneity in other TPM1 HCM-variants,4–11 with SCD episodes in individuals with early onset HCM. In contrast, there were no major CV events among our carriers with early onset HCM; however, there was an SCD episode in a nongenotyped first-degree relative at age 19 years (note that an additional relevant pathogenic MYH7 variant was identified in this pedigree #7), and another case of SCD was reported in a nongenotyped second-degree relative at the age of 21 years. No HCM was reported in either carrier.
In this study, we report a significant number of unaffected carriers at advanced ages in our pedigrees, and also in the general population, which suggests incomplete penetrance. This late onset of the disease and the incomplete penetrance of the variant could represent a challenge when attempting to demonstrate familial cosegregation of a rare variant, such as TPM1 p.Arg21Leu, requiring the grouping of a larger number of families, as we have done here.
Homozygous carriers and additional genetic variantsAlmost 18% of the carriers had a second genetic variant, a percentage higher than that described in the literature (5%) for HCM.2 Previous studies have demonstrated that the number of genetic variants may be a determinant of disease severity.30,31 However, our data demonstrate that the presence of a second variant is not necessarily associated with a more severe phenotype. The majority of carriers with an additional genetic variant had mild phenotypes or were unaffected, which could be explained by the presence of 2 variants with late or incomplete penetrance. The clinical interpretation of genetic findings requires consideration of the clinical features and penetrance described specifically for each of the identified variants.
Further, we could also hypothesize that the presence of an additional genetic variant would be necessary for the penetrance (disease manifestation) of a rare variant identified in the general population, but it was not possible to verify this association statistically in our population.
Several studies have associated sarcomere variants in homozygosis to more severe presentations in HCM than when these variants are identified in simple heterozygozity.32,33TPM1 p.Arg21Leu homozygous carriers in our study showed in general a more pronounced phenotype than simple heterozygous carriers (especially considering the existence of moderate-severe diastolic dysfunction, and ventricular arrhythmias); however, these homozygous carriers showed late onset disease manifestations, supporting the view that this variant is probably not be associated with a life threatening phenotype.
TPM1 p.Arg21Leu as a founder variantThe geographical distribution of the carrier families showed a prevalent concentration in the western part of the Iberian Peninsula, especially in the Spanish regions of Galicia and Extremadura, and northern Portugal (figure 1). These territories share common historical and geopolitical factors dating back to the 11th century, or even earlier.34 Four index cases were identified in collaborating hospitals located outside these zones, but the origin of the families was reported in the founder region. Furthermore, the identification of homozygous carriers with nonconsanguineous progenitors would also reflect the greater prevalence of the variant in specific regions.
A recent study has already described a founder variant for the GLA gene in the same northern Portuguese region, where we also identified a high number of families carrying the TPM1 p.Arg21Leu variant.35 The authors demonstrated that it was related to cultural and socioeconomic particularities (ie, a high level of endogamy due to marriages within the same social stratum) existing since the 17th until 19th century in this region. They considered that social factors and the late penetrance of the variant perpetuated disease transmission in that region until the contemporary era. These characteristics could be hypothesized to explain the distribution observed for the TPM1 p.Arg21Leu variant.
The presence of our variant exclusively in Latin individuals from gnomAD and TOPMed populations could reflect a possible common ancestor from Spain and Portugal, given that these countries had a central role in the colonization of Latin America. In comparison, another TPM1 variant, p.Asp175Asn, described as a founder effect in Finland, is reported to be found predominantly in Finnish European individuals in the gnomAD database.
LimitationsThis genotype-phenotype correlation study has some potential limitations. First, no clinical or genetic evaluation was reported in a high number (n=106) of first-degree relatives identified in the pedigrees. This may be related to the low clinical impact of the variant and the risk perception of carrier families and physicians. Some tests were done only in a limited number of cases, such as cardiac magnetic resonance imaging, limiting our ability to assess additional risk markers. Further, the Kaplan-Meier curve presented in this study showing age at diagnosis should not be considered the real age at onset of disease expression, but only the moment when the diagnosis was made.
Although we describe a relatively large population carrying the same variant, the numbers are still too small to allow definitive conclusions. In this regard, a comparative study of the TPM1 p.Arg21Leu clinical features vs others genetic variants in this same gene may provide additional evidence for more assertive risk stratification in HCM. Finally, there is a need for further studies to better characterize the founder effect suggested here, such as haplotype studies. However, we believe that the available epidemiological data are sufficient to support a founder effect.
ConclusionsThe availability of a large number of carriers with the same TPM1 variant from a defined geographic area in Galicia/Extremadura/Northern Portugal enabled genotype-phenotype evaluation, which revealed the p.Arg21Leu variant to be pathogenic and associated with late onset HCM, incomplete penetrance, and a generally favorable clinical course.
- –
TPM1 is one of the main HCM genes, accounting for 1% to 5% of cases of the disease.
- –
The available clinical information on TPM1 carriers is relatively scarce in the literature. Therefore, genotype-phenotype correlation studies of populations carrying the same sarcomere HCM causal variant could be the ideal opportunity to better understand the clinical profile and associated prognosis.
- –
This is the largest HCM population ever described with the same TPM1 variant, possibly constituting a founder effect in Portugal and Spain.
- –
TPM1 p.Arg21Leu is a pathogenic HCM variant with late onset/incomplete penetrance and a generally favorable prognosis.
- –
This study could be useful for the clinical interpretation of genetic findings in HCM and for the development of health policies in these regions.
No funding source was applied in this research.
CONFLICTS OF INTERESTA. Lamounier Junior and M. Ortiz report personal fees from Health in Code SL, outside the submitted work. L. Monserrat Iglesias is shareholder of Health in Code SL.
Supplementary data associated with this article can be found in the online version available at https://doi.org/10.1016/j.rec.2021.01.001