
Long QT syndrome is an inherited ion channelopathy that leads to syncope and sudden death. Because of the heterogeneous phenotype of this disease, genetic testing is fundamental to detect individuals with concealed long QT syndrome. In this study, we determined the features of a family with 13 carriers of the KCNH2-H562R missense mutation, which affects the pore region of the HERG channel.
MethodsWe identified the KCNH2-H562R mutation in a 65-year-old man with a prolonged QTc interval who had experienced an episode of torsade de pointes. Subsequently, a total of 13 mutation carriers were identified in the family. Carriers (age 48 [26] years; 46% males) underwent clinical evaluation, electrocardiography and echocardiography.
ResultsThe mean (standard deviation) QTc in carriers was 493 (42) ms (3 [23%] showed normal QTc); 6 (46%) had symptoms (4, syncope; 1, sudden death; 1, aborted sudden death [proband]). While under treatment with beta-blockers, 11 of 12 carriers (92%) remained asymptomatic at 5 years of follow-up (1 patient required left cardiac sympathectomy). The QTc shortening with beta-blockers was 50 (37) ms. There was 1 sudden death in a patient who refused treatment.
ConclusionsFamily study is essential in the interpretation of a genetic testing result. This article describes the heterogeneous and variable phenotype of a large family with the KCNH2-H562R mutation and highlights the role of genetic study for the appropriate identification of at-risk individuals who would benefit from treatment.
Keywords
Congenital long QT syndrome (LQTS) is an inherited channelopathy characterized by a prolongation of ventricular repolarization, which is exhibited as a prolonged QT interval on the electrocardiogram (ECG). This exposes patients to syncope and sudden death by torsade de pointes that degenerate into ventricular fibrillation. To date, more than 700 mutations have been identified in 13 LQTS genes, with types 1 to 3 LQTS being the most frequent genotypes.1 Long QT syndrome type 2 (LQT2) is associated with the human ether-a-go-go-related gene (HERG; KCNH2; Kv11.1) loss-of-function mutations,2 and represents 30% to 45% of patients with a positive genotype for LQTS.1HERG gene encodes the voltage-gated potassium channel α-subunit underlying the rapid component of the cardiac delayed rectifier K+ current. Each α-subunit consists of 6 α-helical transmembrane segments (S1-S6) where the channel pore is between S5 and S6.2KCNH2 mutations in the pore-loop region, which is responsible for forming the ion conduction pathway of the channel, have been shown to be associated with an increased risk for arrhythmic events.3,4 Moreover, the natural history of the disease differs according to sex and age5 and frequently there is a variable gene expression in family members that share the same mutation. Furthermore, the information about the genotype-phenotype correlation is usually based on isolated cases and small pedigrees. Thus, the aim of this study was to describe the cardiac events, their triggers, and the ECG characteristics of a large family with LQT2, and to evaluate the response to beta-blocker therapy.
METHODSStudy PopulationThe proband was a 65-year-old man who presented with an episode of polymorphic ventricular tachycardia after 3 days of treatment with a macrolide. The ECG after torsade de pointes showed a QTc interval of 600ms. A pedigree was taken in the patient interview and relatives were invited to undergo screening. The evaluation of the proband and his relatives included medical history, physical examination, 12-lead ECG, the Schwartz score,6 and 2-dimensional and Doppler echocardiography. Blood samples were taken for genetic analysis and the patient and his relatives gave written consent. The study was approved by the local ethics committee.
The QT interval was measured in II and V5 or V6, and was corrected by Bazett's formula (QTc = QT/RR0.5). We considered long QTc interval to be a value > 450ms in men or > 460 in women.7According to the recently published expert consensus on inherited primary arrhythmia syndromes,8 LQTS was diagnosed in the proband by the presence of a LQTS risk score ≥ 3.56 and a corrected QT interval ≥ 500ms in repeated ECGs. After the identification of the KCNH2-H562R mutation in the proband, the diagnosis in relatives was established based on the genetic study of the mutation.
Genetic StudyGenomic DNA was extracted from peripheral blood samples using standard protocols. All exons of KCNQ1, KCNH2, SCN5A were amplified with intronic primers and sequenced in both directions using BigDye v1.1 chemistry in an ABI3130 analyzer (Applied Biosystems; Foster City, California, United States). The sequence for each gene was compared with the reference sequence in the National Center for Biotechnology Information sequence database. The pathogenicity of the new variant was evaluated using in silico software.
Statistical AnalysisWe used the SPSS statistical program version 15.0 for the analysis (SPSS; Chicago, Illinois, United States). Continuous variables were tested for normal distribution using the Kolmogorov-Smirnov test. Normally distributed variables are expressed as mean (standard deviation) and qualitative variables as absolute values and percentages. Mann-Whitney's U test was used to objectify the presence of differences between the carriers and noncarriers of KCNH2-H562R. The QTc shortening under treatment with beta-blockers was evaluated using the Wilcoxon test and the effect of different types of beta-blockers using the Kruskal-Wallis test. P values < .05 were considered statistically significant.
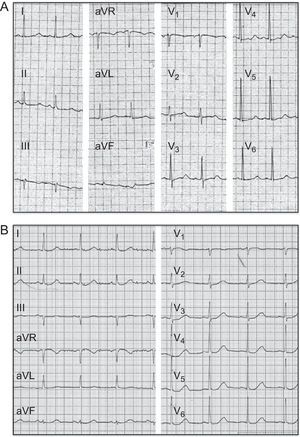
RESULTSClinical HistoryThe index patient (II.2) was a 65-year-old Caucasian man that was admitted to our hospital because of a syncope episode while walking. The patient had been treated with clarithromycin for 3 days due to a respiratory infection. He reported several episodes of syncope since the age of 30 years, but had never been studied for this reason. A blood test showed a mild hypokalemia (K+ 3.1 mEq/L), with no other abnormalities. While he was at the emergency observation area, he experienced an episode of polymorphic ventricular tachycardia that degenerated into ventricular fibrillation and required electric cardioversion. The ECG postcardioversion revealed a QTc interval of 600ms (Figure 1A). Subsequently, clarithromycin was discontinued and the hypokalemia was corrected, as both produce abnormal prolongation of the QT interval. However, the QTc interval was still clearly prolonged (520ms), so treatment with bisoprolol was initiated.

Under treatment with a medium dose of bisoprolol, the QTc was reduced to 490ms (Figure 1B). However, the patient had symptomatic sinus bradycardia and multiple orthostatic presyncopal episodes. Thus, we decided to implant a DDD-R pacemaker (the patient refused an implantable cardioverter defibrillator) to increase the heart rate and the dose of beta-blockers, with the purpose of reducing the QTc interval and controlling the sinus bradycardia symptoms. After atrial pacing and maximum dose of bisoprolol, QT was reduced to 470ms. Subsequently, he has remained asymptomatic and had no arrhythmic events in the 24-h Holter ECG or the exercise test at annual follow-up visits during a 5-year follow-up period.
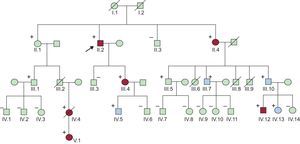
The patient's pedigree was taken and his first degree relatives were screened (Figure 2). Genetic analysis of cardiac ion channel genes was performed in the proband. This analysis revealed that the index case was heterozygous for the KCNH2-H562R missense mutation, being diagnosed with LQT2.
. In blue are the carriers that remained asymptomatic despite a prolonged QTc interval, and in red the patients who presented symptoms (syncope, sudden death, aborted sudden death).")
Pedigree. Patients III.6, III.8 and III.9 died before reaching 1 year of age. Carriers of the mutation are marked with a (+). In blue are the carriers that remained asymptomatic despite a prolonged QTc interval, and in red the patients who presented symptoms (syncope, sudden death, aborted sudden death).
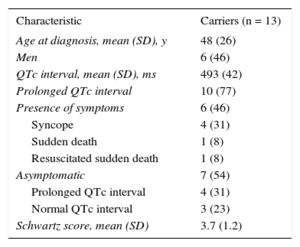
Twenty-four family members (Figure 2) were screened for the mutation previously found in the proband, and 12 relatives (50%) were carriers of the KCNH2-H562R mutation. Demographic characteristics, QTc intervals, and events in mutation carriers are shown in Table 1.
Clinical and Electrocardiographic Characteristics of KCNH2-H562R Carriers
| Characteristic | Carriers (n = 13) |
|---|---|
| Age at diagnosis, mean (SD), y | 48 (26) |
| Men | 6 (46) |
| QTc interval, mean (SD), ms | 493 (42) |
| Prolonged QTc interval | 10 (77) |
| Presence of symptoms | 6 (46) |
| Syncope | 4 (31) |
| Sudden death | 1 (8) |
| Resuscitated sudden death | 1 (8) |
| Asymptomatic | 7 (54) |
| Prolonged QTc interval | 4 (31) |
| Normal QTc interval | 3 (23) |
| Schwartz score, mean (SD) | 3.7 (1.2) |
SD, standard deviation.
Data are expressed as mean (standard deviation) or No. (%).
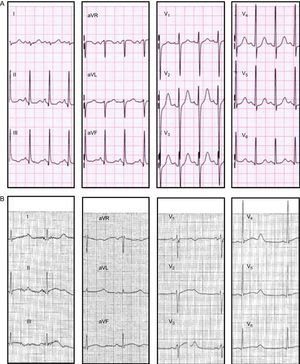
The family history revealed 3 unexplained deaths among children aged less than 1 year (III.6, III.8, III.9) (ECGs were not available and genetic study was not performed). Among the 13 carriers of the mutation, 6 patients (46%, including the index case) had cardiac symptoms (Table 2). Patient IV.4 had been diagnosed with epilepsy due to several nocturnal convulsive episodes. She showed a wide variation of the QTc interval in previous ECGs (QTc from 430ms to 480ms) and her cardiologist prescribed beta-blocker therapy, but she refused treatment. One night the phone rang while she was sleeping and she lost consciousness shortly after the conversation started; 6 years later, she died suddenly during sleep. Her father (III.2), who had to be an obligate carrier of the mutation, had died suddenly at home 15 years before at the age of 35 (no autopsy was performed). The daughter of patient IV.4 (patient V.1), had several episodes of syncope at rest despite treatment with propanolol (QTc 480ms; Figure 3A), requiring a left cardiac sympathetic denervation at the age of 6 years because she was considered too young for an implantable cardioverter defibrillator.
Events in Family Members Carrying the KCNH2-H562R Mutation
| Patient | Age at first evaluation, years | QTc at first evaluation, ms | Symptoms |
|---|---|---|---|
| II.4 | 83 | 508 | Syncope at rest |
| III.4 | 53 | 490 | Syncope at rest |
| IV.4 | 23 | 455 | Convulsive episodes (diagnosed with epilepsy); syncope triggered by an acoustic stimulus; sudden death |
| IV.12 | 20 | 600 | Syncope at rest |
| V.1 | 3 | 488 | Syncope at rest |
. B: patient IV.12 (QTc, 600 ms). Both reported several episodes of syncope at rest.")
Currently, all living carriers (n = 12) are under treatment with beta-blockers, and only the aforementioned patient V.1 has shown cardiac symptoms during a 5-year follow-up period.
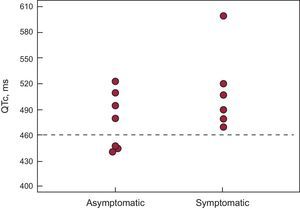
Electrocardiographic Study of Mutation CarriersThe typical notched T waves on the ECG were observed in 3 (23%) patients (Figure 3B), with broad T waves being the predominant morphology in the other patients. The QTc interval was longer in mutation carriers than in non-carrier relatives QTc, 493 [42] ms vs 418 [14] ms; P < .001). QTc was prolonged in symptomatic rather than in asymptomatic carriers, although the difference was not statistically significant (QTc, 517 [57] ms vs 477 [33] ms; P = .28) (Figure 4). QTc interval was normal in 23% of carriers (n = 3), with all of them being asymptomatic. Females had a higher QTc than males but this difference was not statistically significant (QTc, 508 [57] ms vs 481 [21] ms; P = .23).

While the results of the genetic study were available, stress testing was done in those family members with a normal QTc to identify probable carriers. In the 3 individuals with normal QTc who were later found to be carriers of the mutation, the QTc interval showed similar behavior with exercise: during the first minute the QTc increased in 25 (5) ms, then progressively shortened during the rest of the exercise, and finally in the third minute of the recovery phase the QTc increased by 25 (10) ms. This pathological behavior allowed an early initiation of beta-blockers in these patients.
All carriers received beta-blockers (except IV.4, as noted above). Of these, 5 (42%) were initiated on bisoprolol, 4 (33%) on metoprolol, and 3 (25%) on propranolol (1 patient was excluded from the analysis due to ventricular pacing). The mean QTc shortening with beta-blockers was 50 (37) ms (n = 11; QTc, 493 [46] ms vs 442 [19] ms; P = .002). The QTc shortening was 44 (25) ms with bisorpolol, 47 (23) ms with metoprolol and 63 (73) ms with propranolol (P = .9).
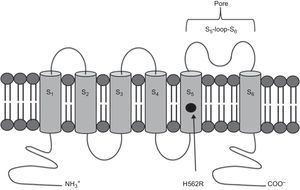
Genetic AnalysisThe c.1685A>G, p.H562R (rs199472922) heterozygous variant was detected in exon 7 of the KCNH2 gene. This variant causes an amino acid change from histidine 562 to arginine, with this replacement taking place in the S5 transmembrane region of the protein (Figure 5). The amino acid involved is located in a highly conserved region between the species and paralogue genes. A bioinformatic study of the mutation was performed using “Mutation Taster”, resulting in a pathogenic variant with P = .99 (P indicates the prediction value; values close to 1 indicate high reliability of the prediction made).
DISCUSSION
In the present study, we describe for the first time the phenotype of the KCNH2-H562R missense mutation in a large family with LQTS. Our clinical data show a proband with a history of syncope that suffered a resuscitated sudden death in the setting of hypokalemia and clarithromycin therapy.
Implications of HERG Mutations Located in the Pore RegionIn humans, KCNH2 is located on chromosome 7q35-36, and the coding region comprises 16 exons.2 The full length HERG1 subunit is composed of 1159 amino acids and has 6 transmembrane domains (S1–S6). The K+ selective pore is found between transmembrane domain S5 and S6 (amino acid residues 550 through to 650). The mutation that we found in the family studied is located in the S5 domain, being part of the channel pore. In 2002, Moss et al3 published the follow-up which extended over a period of 40 years analyzing 44 different HERG mutations, in which subjects with pore mutations had more severe clinical manifestations and a higher frequency of arrhythmia-related events (74% vs 35%; P < .001). Later, Shimizu et al4 proposed that the S5-loop-S6 region was also associated with the greatest risk in a study including 858 LQT2 patients with 162 different KCNH2 mutations. The reason for this is that pore mutations have a greater negative effect on cardiac delayed rectifier K+ current. However, only a small percentage of KCNH2 mutations have been characterized by a functional in vitro electrophysiological analysis.2,4
The KCNH2-H562R mutation was reported in 2 isolated patients in a study that examined the spectrum of mutations found in 2500 unrelated cases,9 but the authors did not describe the phenotype-genotype relationships of the genetic variants found. In addition, Sharma et al10 showed that the functional analysis of another mutation in the same amino acid (H562P) resulted in a marked reduction of the K+ current in a patient with several syncopal events. All these findings support the pathogenicity of the mutation found in the family described here.
However, assessment of the pathogenicity of an identified genetic variant is sometimes controversial, and is currently more common after the introduction of new genotyping technologies of next-generation sequencing.11,12 With these genotyping techniques, a large number of genes can be analyzed, thereby increasing the problems for interpretation. Thus, recent genome studies have questioned the pathogenicity of up to 17% of suggested diseases causing genetic variants in cardiomyopathies.11,12 In this article, we show how the clinical information and the analysis of cosegregation of the mutation with the disease allowed us to establish the diagnosis.
Genotype-phenotype CorrelationThe ECG study showed a lower frequency of typical notched T waves,13 with only 23% of carriers showing this pattern. In addition, we observed a large variance in phenotype and QTc interval duration in family members sharing the same mutation. This variable penetrance could be partially explained by the coexistence of a single polymorphism on LQT-causing genes and/or unknown genes and/or multiple gene mutations.14
We observed no differences in the duration of the QTc in carriers with and without symptoms. Thus, we could not identify a priori by using the QTc interval value, those subjects who would develop symptoms during the follow-up. In addition, there were 3 asymptomatic carriers that showed a normal QTc. In this regard, it is known that there is an important overlap in the distribution of QTc between otherwise healthy subjects and patients with genetically confirmed LQTS. According to the published literature, 40% of the genotyped LQTS population exhibit QTc values < 460ms.15
Multiple studies have shown the usefulness of QTc response to exercise in patients with LQTS. Long QT syndrome type 1 patients usually have a marked prolongation of QTc during exercise, whereas LQT2 patients have a discreetly prolonged or not prolonged QT interval.16 Later, it was described that the QT interval is prolonged at 2-3min of the recovery phase in patients with LQT2,17,18 a finding also observed in carriers of this family.
Sex has been identified as an important independent factor affecting event risk in LQT2, wherein women display a significantly higher risk of cardiac events than men after the onset of adolescence.5 This is corroborated in our study, in which women had a higher QTc and 4 of the 6 symptomatic carriers (66%) were females.
In patients with LQT2, most cardiac events are associated with an increased sympathetic tone, emotional stress, and/or sudden auditory stimulation, which are the most common triggers for arrhythmia,19 such as in patient IV.4. A study published in 201020 showed that the most important risk factors for arousal-triggered cardiac events were female sex and pore-loop mutations, while those for exercise-triggered events were the presence of nonpore-loop mutations. However, other carriers of the mutation studied showed symptomsat rest. In this context, several studies have reported that about 30% of the cardiac events in LQT2 patients occurred at rest.21 Thus, REM sleep is associated with deep sympathetic activation, which could play an important role in triggering cardiac events in LQT2 patients.
EpilepsyIt is not uncommon for LQTS patients to be diagnosed with epilepsy. Accordingly, the KCNH2 gene was initially discovered in the hippocampus, and its expression was later demonstrated in many regions of the central nervous system. In a study that included 343 LQTS probands, a personal history of seizures was more common in LQT2 than all other subtypes of LQTS combined.22 In the family studied, patient IV.4 had a previous history of pharmacologically treated epilepsy. Some antiepileptic drugs such as phenytoin and other antiepileptic drugs have arrhythmogenic potential because of their cardiac delayed rectifier K+ current blocker potential in vitro. Therefore, an ECG should be performed in all patients with syncope and seizures to rule out LQTS,2 since treatment with antiepileptic drugs may be inadequate and deleterious.
Other TriggersThere are multiple causes of abnormal QT interval prolongation such as myocardial ischemia, cardiomyopathies, hypothermia, hypokalemia, hypocalcemia, hypomagnesemia, autonomic influences, the effects of drugs, and the recently described decrease in epicardial temperature.23 Hypokalemia and clarithromycin may have been the triggering factors in the index patient. Some studies have recently demonstrated a reduction in QTc interval in LQT2 patients with a combination of potassium supplementation and aldosterone-inhibition. We recommended treatment with potassium supplements in the proband and other carriers with hypokalemia or potassium levels toward the lower limit of normality. Furthermore, there is a wide array of drugs that prolong the QT interval and/or induce polymorphic ventricular tachycardia.24 In this context, prolonged QT interval and ventricular arrhythmias have been reported in patients treated with erythromycin because this drug produces an inhibition of the cardiac delayed rectifier K+ current channel.25 Similar to our index patient, several cases of QT prolongation and torsades de pointes after clarithromycin administration have been published. Since the structure of clarithromycin is similar to that of erythromycin, the arrhythmogenic properties should be similar in both.26 Other macrolides such as roxithromycin and azithromycin appear to be less arrhythmogenic in comparative in vitro studies.27
TreatmentTherapy for LQTS is directed toward reducing the incidence of syncope and sudden death. The standard treatment includes beta-blockers, sometimes used in combination with an implantable cardioverter defibrillator.28 The effectiveness of beta-blockers was documented in a large study that included 869 LQTS patients, where cardiac events were reduced in probands from 0.97 to 0.31 and in affected family members from 0.26 to 0.15 events per year.29 In our cohort, with the exception of the girl V.1, we observed no events after the initiation of beta-blockers, observing a significant reduction in the mean QTc interval.
The protective effect of beta-blockers in the LQTS population is not uniform because its mechanism of action is probably related to the attenuation of adrenergic-mediated triggers. Prior data suggest that there is a high rate of cardiac events among patients with LQT2 and LQTS type 3 treated with beta-blockers.30 A later study published in 2011 suggests that within the LQT2 population there is a trigger-specific response to beta-blocker therapy, so that LQT2 patients experience a very low rate of exercise-triggered events during beta-blocker therapy, but have a high rate of arousal (more abrupt changes in heart rate than exercise) and nonexercise/nonarousal (not associated with sympathetic activation) triggered cardiac events despite recommended therapy.31 However, 13% and 17% of the patients who experienced a first arousal- and nonexercise/nonarousal-triggered event, respectively, experienced a subsequent exercise-triggered event.32 Migdalovich et al5 reported that medical therapy with beta-blockers was associated with a 61% reduction in the risk of sudden death in a population of 1166 LQT2 patients. A recent study comparing the efficacy of commonly used beta-blockers showed that propranolol had a higher QTc shortening effect than metoprolol and nadolol.32
The only patient that remained symptomatic underwent left cardiac sympathetic denervation. In a study involving 147 LQTS patients, left cardiac sympathetic denervation reduced cardiac events by approximately 90%.33
Risk StratificationThe most widely accepted risk stratification of LQTS is based on published mortality rates, and classifies patients in high (history of aborted and/or ECG-documented episodes of polymorphic ventricular tachycardia), intermediate (prior syncope and/or QTc > 500ms), and low (no prior syncope and QTc ≤ 500ms)29 risk. However, a recent study showed a different association between mutation characteristics and time-dependent differences in the clinical course of LQT2 patients. Therefore, after the onset of adolescence, women with and without high-risk mutations show increased risk for life-threatening cardiac events, whereas the risk in men only increased among carriers of the higher-risk pore-loop mutations.5 Therefore, the current trend is toward a combined assessment of clinical and mutation-specific data to improve risk stratification for life-threatening cardiac events in LQT2.
CONCLUSIONSThis article describes the clinical manifestations and QTc interval values of the KCNH2-H562R missense mutation for the first time in a large family of carriers. We corroborate previous data about the high risk associated with pore mutation and focus on the clinical and electrocardiographic heterogeneity of this channelopathy. Within this framework, our study highlights the fundamental role that genetic testing has acquired in deciding which individuals are appropriate for treatment. Finally, we emphasize the current importance of cosegregation analysis of the mutation with the disease in the family, particularly in an era of technological development when the techniques of next-generation sequencing are acquiring an increasing role.
CONFLICTS OF INTERESTNone declared.