X-linked cardiac valvular dysplasia is a rare form of male-specific congenital heart defect mainly characterized by myxomatous degeneration of the atrioventricular valves with variable hemodynamic consequences. It is caused by genetic defects in FLNA-encoded filamin A, a widely expressed actin-binding protein that regulates cytoskeleton organization. Filamin A loss of function has also been associated with often concurring neurologic and connective tissue manifestations, with mutations in the first half of the Rod 1 domain apparently expressing the full cardiac phenotype. We contribute to previous genotype-phenotype correlations with a multidisciplinary approach in a newly-described family.
MethodsCardiologic, dysmorphologic, and genetic evaluation of available members were complemented with transcriptional and X-chromosome inactivation studies.
ResultsA novel FLNA mutation c.1066-3C>G cosegregated with a male-expressed, apparently isolated, cardiac phenotype with no skewed X-inactivation pattern in female carriers. This variant was shown to result in an in-frame deletion of 8 amino acid residues near the N-terminal region of the protein.
ConclusionsA nonimprinted, partial loss of function of filamin A proximal Rod 1 domain seems to be the pathogenetic mechanism of cardiac valvular dysplasia, with some cases occasionally expressing associated extracardiac manifestations.
Keywords
Multivalvular heart disease is the combination of stenotic and/or regurgitant lesions of 2 or more cardiac valves. It is a highly prevalent clinical condition among patients with an underlying valvular disease, affecting approximately 20% of patients with congenital valve defects and 15% of patients undergoing valvular surgery.1 In the PARTNER trials, the incidence of concomitant moderate-to-severe mitral regurgitation in patients with severe aortic stenosis was approximately 20%, and the incidence of moderate-to-severe tricuspid regurgitation was 27%; however, many of these valvular heart diseases were also secondary to hemodynamic alterations, ischemic heart disease, and ventricular dysfunction.1 The complexity of determining the etiology of multivalvular disease hampers diagnosis and patient management.
X-linked cardiac valvular dysplasia (XCVD, MIM 314400), also known as polyvalvar myxomatous valve degeneration, is a rare disorder caused by mutations in the filamin A gene (FLNA). These genetic defects underlie early-onset, progressive myxomatous mitral and tricuspid valve deterioration that presents as valve thickening and dysfunction followed by secondary chamber remodeling and ultimately heart failure. Only a few cases and families have been described to date with this genetic disorder, some of whom were retrospectively diagnosed.2
We report a family with 5 individuals confirmed or suspected to have XCVD due to a novel mutation, c.1066-3C>G, in intron 7 of FLNA. This variant abolishes the canonical splice acceptor site, resulting in the in-frame deletion of 8 amino acids of the proximal Rod 1 domain. Thus, this mutation may result in partial loss of function of filamin A, consistent with most previously reported XCVD-associated variants. We review previous XCVD cases and speculate about their different phenotypes, depending on the functional effect, the impact of the variants, and the filamin A topology.
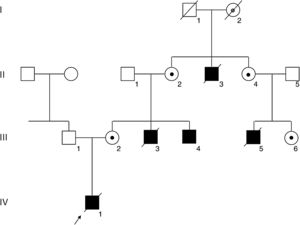
PatientsA complete pedigree of the family is shown in Figure 1.
. The FLNA mutation was confirmed in the living affected male III-4 and in carrier females II-2, II-4, III-2, and III-6, depicted with a black dot. Obligate carrier I-2 is depicted with a gray dot.")
Pedigree of the family. Squares and circles depict male and female members, respectively. Affected members are shown in black, and the deceased members are crossed with a slash. An arrow indicates the index patient (IV-1). The FLNA mutation was confirmed in the living affected male III-4 and in carrier females II-2, II-4, III-2, and III-6, depicted with a black dot. Obligate carrier I-2 is depicted with a gray dot.
Index case IV-1. This patient was diagnosed prenatally with abnormally thick cardiac valves by ultrasound and fetal echocardiography ( and ). There was moderate mitral regurgitation and severe tricuspid regurgitation leading to an aneurysmatic dilatation of the right atrium. The pulmonary and the aortic valves were also dysplastic and apparently small. He was born at term (39 weeks) with a birth weight of 3950g and severe neonatal distress, requiring extracorporeal membrane oxygenation. On the first day of life, echocardiography showed a large dilatation of the right atrium, severe tricuspid insufficiency due to a dysplastic valve, moderate mitral insufficiency with a dysplastic mitral valve, a 6-mm atrial septal defect with right-left shunt, dilatation of the right ventricle, and left ventricular hypertrophy. The aortic and pulmonary valves were also dysplastic with regurgitation. The pulmonary artery was moderately dilated and the ductus was large with bidirectional flow, predominantly left-right. There was also a slight pericardial effusion. The clinical behavior was of a patient with hypoplastic left heart with functional aortic atresia and a patent ductus dependency. No features of a connective tissue disorder were reported on clinical examination. He underwent cardiac surgery to repair both valves but died intraoperatively. Neither autopsy nor previous brain ultrasound revealed no neuronal migration disorder. Standard karyotype was normal.
Affected relative III-4. This is a 27-year-old man with a congenital heart defect. He was born at term with respiratory distress. A heart murmur was detected at 2 months of age. Postnatal echocardiography confirmed that all valves were thickened and dystrophic, with moderate tricuspid regurgitation, mild mitral and pulmonary regurgitation, and mild aortic incompetence (). Clinical examination showed no joint hypermobility or skin hyperextensibility. Brain magnetic resonance imaging showed no neuronal migration disorder.
Affected relative III-3. This individual was born at term after an apparently normal pregnancy and delivery. Soon after birth, he developed cyanosis and respiratory distress and died on the first day of life, more than 30 years ago. No other data are available.
Affected relative III-5. This child was born preterm (37 weeks) after a normal pregnancy and delivery. He remained in neonatal intensive care for 2 weeks due to prematurity and abnormal feeding. He underwent cardiac surgery in another country and, after 2 months of indolent clinical course, died after a respiratory illness. The family reported that the neonate had abnormal cardiac valves with cavity dilatation and large heart.
Affected relative II-3. This neonate was born at term, in a home birth more than 60 years ago. The only information reported by the family is that the boy had cardiac murmurs and failure to thrive. No other data are available.
Echocardiography was performed on asymptomatic female relatives (III-2, II-2, II-4, III-6), yielding normal results in all of them except for a 58-year-old woman, II-2, who showed mild pulmonary regurgitation. On clinical examination, there was no joint hypermobility or skin hyperextensibility. No brain magnetic resonance imaging or ultrasound was performed.
METHODSGenetic AnalysisAll participants provided informed consent for the studies conducted and ethical approval was obtained from the respective institutions. The FLNA coding sequence and intron-exon boundaries (NM_001456.3) were amplified and sequenced using genomic DNA extracted from peripheral blood lymphocytes of the mother of the proband (III-2) as no sample could be obtained from the index case. Familial segregation studies were subsequently performed.
RNA StudiesPeripheral blood from the living affected male, III-4, was collected in PAXgene blood RNA tubes (PreAnalytiX GmBH, Qiagen NV/Becton Dickenson & company) for RNA extraction. RNA was extracted using the blood RNA kit (PreAnalytiX GmBH) and complementary DNA (cDNA) synthesis was performed using the High Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific), both according to the manufacturer's instructions. cDNA was amplified using an amplicon that contains FLNA exons 6-9 (NM_001456.3), using the oligonucleotides: 5¿ -ACACCAGGAGGAGGCAAAAG-3¿ located across the exon 6 and 7 junction, and 5¿-GCTCTACCGTGCCCTTCTGT-3¿ in exon 9. The products were visualized by electrophoresis and subsequently sequenced.
X-chromosome InactivationX-chromosome inactivation pattern was determined using genomic DNA from the lymphocytes of available female carriers by means of an indirect methylation quantitative assay in the androgen receptor gene, as previously described.3
Bioinformatic AnalysisVariants with a minor allele frequency below 0.1% were filtered according to available data in the gnomAD browser.4In silico pathogenicity predictions were determined using Alamut Visual V2.9.0 including the following splicing simulation tools: SpliceSiteFinder-like, MaxEntScan, NNSPLICE, GeneSplicer and Human Splicing Finder. Variant classification was made according to the ACMG standards and guidelines for the interpretation of sequence variants.5
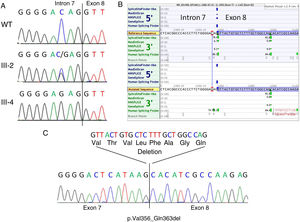
RESULTSA heterozygous intronic variant, c.1066-3C>G, was detected in FLNA in the mother of the index case (III-2, Figure 2A), at position -3 from the splicing acceptor site in intron 7. This variant was absent in more than 177600 alleles in the gnomAD database. Family members II-2, II-4, III-4, and III-6 also tested positive for the same variant, confirming segregation in all available affected and carrier members and denoting I-2 as an obligate carrier (Figure 1).
, heterozygous female carrier (III-2, middle panel) and hemizygous affected male (III-4, lower panel). The intron 7-exon 8 boundary is indicated. B: In silico splicing predictions showing the consequence of the variant on the acceptor site of intron 7. Note that a strong cryptic acceptor site is located 24bp downstream in exon 8. C: cDNA sequencing of patient III-4 showing that indeed the canonical splice acceptor site in intron 7 is abolished in the mutant cDNA and that the alternative downstream acceptor site in exon 8 is used. This results in the in-frame deletion of 24bp (8 amino acids).")
Genetic studies. A: Genomic sequencing chromatogram showing the c.1066-3C>G variant in a normal control (WT, upper panel), heterozygous female carrier (III-2, middle panel) and hemizygous affected male (III-4, lower panel). The intron 7-exon 8 boundary is indicated. B: In silico splicing predictions showing the consequence of the variant on the acceptor site of intron 7. Note that a strong cryptic acceptor site is located 24bp downstream in exon 8. C: cDNA sequencing of patient III-4 showing that indeed the canonical splice acceptor site in intron 7 is abolished in the mutant cDNA and that the alternative downstream acceptor site in exon 8 is used. This results in the in-frame deletion of 24bp (8 amino acids).
The canonical splice site acceptor in intron 7 was totally inactivated in the mutated sequence according to SpliceSiteFinder-like, MaxEntScan and GeneSplicer, and significantly weakened (76.17 vs 86.47) as predicted by Human Splicing Finder (Figure 2B). According to in silico splicing predictions, a cryptic acceptor site exists 24bp downstream within exon 8, potentially resulting in the rescue of the protein reading frame (p.Val356_Gln363del). This prediction was confirmed by RNA studies from the peripheral blood of patient III-4 (Figure 2C).
The X-chromosome inactivation pattern was determined for female carriers III-2, II-2, and III-6. They all showed a random pattern of inactivation (50:50, 45:55 and 44:56, respectively). Female carrier II-4 was uninformative for the polymorphism employed in this assay.
DISCUSSIONBoth syndromic and isolated forms have been described for hereditary valve disease. Connective tissue disorders such as Marfan syndrome and more rarely Loeys-Dietz syndrome, show mitral valve prolapse as well as other valve dysfunctions as a part of a wider vascular, ocular, and skeletal clinical spectrum.6 Ehlers-Danlos syndrome, especially the autosomal recessive cardiac valvular form, caused by mutations in COL1A2, also present with left-side valve manifestations in different degrees of severity.7
The XCVD was the first nonsyndromic valve dystrophy to be genetically characterized by linkage analysis showing defects in filamin A as the underlying etiology.8 It is distinguished by early-onset bilateral multivalve dysfunction with predominant left involvement, likely due to the higher hemodynamic stress associated with mitral and aortic valves.2 It is inherited in an X-linked, apparently recessive manner that typically shows complete penetrance in males and a subclinical reduced expression among female carriers.8
However, FLNA mutations have also been associated with a wide variety of distinct genetic diseases including periventricular nodular heterotopia (PVNH, MIM 300049),9 FG syndrome type 2 (MIM 300321), neuronal intestinal pseudo-obstruction (MIM 300048), terminal osseous dysplasia (MIM 300244), and the otopalatodigital spectrum disorders otopalatodigital types I (MIM 311300) and II (MIM 304120), Melnick-Needles syndrome (MIM 309350) and frontometaphyseal dysplasia (MIM 305620).10 It has been proposed that total and partial loss of filamin A function result in neuronal migration disorders and valvular dystrophies, respectively, whereas a gain of function mechanism seems to be associated with skeletal dysplasias.10 Nevertheless, exceptions to this model have been observed in phenotypic outcome, tissue expression, and expected male lethality.11–13 Mixed phenotypes have also been described,14 including transcript-dependent opposite functional effects of single variants.15,16
Severe mitral and pulmonary valve defects have been occasionally observed in patients with PVNH.17,18 Before these reports, bicuspid aortic valve, aortic root dilatation, and patent ductus arteriosus were the only cardiovascular manifestations that had been associated with PVNH. Moreover, an Ehlers-Danlos syndrome-like phenotype including joint hypermobility and skin hyperlaxity has also been found in patients with FLNA-related PVNH.19–21 Valvular defects and connective tissue disorders are both included in a single extracellular matrix-related etiologic spectrum and, in fact, both conditions have been concurrently observed in filamin A patients.22–25 Unlike skeletal dysplasias, all these defects are apparently associated with a complete or partial loss of function of filamin A and may be part of a clinical continuum. Thus, FLNA-related XCVD should not be considered a nonsyndromic form of valvular dystrophy, but rather a pleiotropic disorder with variable expressivity, possibly related to the functional alteration and impact of the mutation.
Indeed, among previously reported cases with XCVD, connective tissue manifestations and even PVNH seem to be common in patients in whom those features were examined (Table). Marfanoid skeletal features are routinely tested when congenital valvular defects are found, whereas PVNH, even in the absence of epilepsy or seizures, can only be ruled out by brain magnetic resonance imaging. On the other hand, the only families apparently showing PVNH have unspecified FLNA mutations not revealing the affected domain or the impact of the mutation (Table).
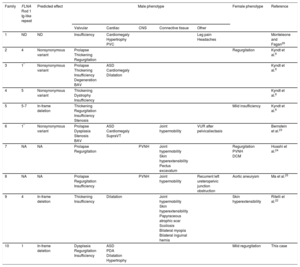
Summary of Cardiac and Extracardiac Phenotypes and Genetic Findings in Patients With X-linked Cardiac Valvular Dysplasia
| Family | FLNA Rod 1 Ig-like repeat | Predicted effect | Male phenotype | Female phenotype | Reference | ||||
|---|---|---|---|---|---|---|---|---|---|
| Valvular | Cardiac | CNS | Connective tissue | Other | |||||
| 1 | ND | ND | Insufficiency | Cardiomegaly Hypertrophy PVC | Leg pain Headaches | Monteleone and Fagan26 | |||
| 2 | 4 | Nonsynonymous variant | Prolapse Thickening Regurgitation | Regurgitation | Kyndt et al.8 | ||||
| 3 | 1* | Nonsynonymous variant | Prolapse Thickening Insufficiency Degeneration BAV | ASD Cardiomegaly Dilatation | Kyndt et al.8 | ||||
| 4 | 5 | Nonsynonymous variant | Thickening Dystrophy Insufficiency | Kyndt et al.8 | |||||
| 5 | 5-7 | In-frame deletion | Thickening Regurgitation Insufficiency Stenosis | Mild insufficiency | Kyndt et al.8 | ||||
| 6 | 1* | Nonsynonymous variant | Prolapse Dysplasia Stenosis BAV | ASD Cardiomegaly SupraVT | Joint hypermobility | VUR after pelvicaliectasis | Bernstein et al.23 | ||
| 7 | NA | NA | Prolapse Regurgitation | PVNH | Joint hypermobility Skin hyperextensibility Pectus excavatum | Regurgitation PVNH DCM | Hoashi et al.24 | ||
| 8 | NA | NA | Prolapse Regurgitation Insufficiency | PVNH | Joint hypermobility | Recurrent left ureteropelvic junction obstruction | Aortic aneurysm | Ma et al.25 | |
| 9 | 4 | In-frame deletion | Thickening Insufficiency | Dilatation | Joint hypermobility Skin hyperextensibility Papyraceous atrophic scar Scoliosis Bilateral myopia Bilateral inguinal hernia | Skin hyperextensibility | Ritelli et al.22 | ||
| 10 | 1 | In-frame deletion | Dysplasia Regurgitation Insufficiency | ASD PDA Dilatation Hypertrophy | Mild regurgitation | This case | |||
ASD, atrial septal defect; BAV, bicuspid aortic valve; CNS, central nervous system; DCM, dilated cardiomyopathy; NA, not available; ND, not determined; PDA, patent ductus arteriosus; PVC, premature ventricular contractions; PVNH, periventricular nodular heterotopia; SupraVT, supraventricular tachycardia; VUR, vesicoureteral reflux.
Nevertheless, the individuals reported in our study have no extracardiac, neurologic, skeletal, or cutaneous manifestations. The index patient was born with an atrial septal defect, a previously reported malformation in association with defects in filamin A. Notably, the 2 other patients with XCVD and this cardiac defect carried both the same mutation, also in Ig-like repeat 1 within the Rod 1 domain (Table). Vascular defects present in our index patient have also been described in patients with PVNH, and in general, cardiovascular defects are common in FLNA-associated skeletal dysplasias.14
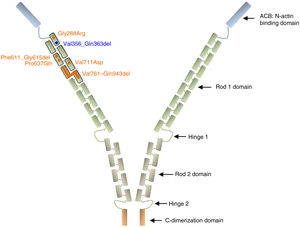
Filamin A is a ubiquitous protein that stabilizes actin filaments and links them to membrane glycoproteins. It has also been reported to interact with many other proteins such as integrins, transmembrane receptor complexes, and second messengers, with a suggested role in signal transduction. Structurally, the conserved actin-binding domain at the N-terminus of the protein sequence is followed by 24 homologous Ig-like repeats of 96 amino acid residues each. This main structure is separated by 2 hinge domains into Rod 1 (repeats 1–15) and Rod 2 (repeats 16–23), which are followed by the C-terminal dimerization domain (repeat 24)27,28 (Figure 3).

Schematic representation of the filamin A protein. Nonsynonymous variants are represented with circles and intragenic deletions with bars. All XCVD reported changes are depicted in orange except the one described in this report, which is shown in blue. Note that all alterations are clustered in the first 7 Ig-like repeats of the Rod 1 protein domain. References for each variant are cited in the text.
Among the 9 previously described families with XCVD, mutations in FLNA were reported in 8 of them (patients of the family described by Monteleone and Fagan in 1969 were not tested).26 In 2 of the 8 FLNA-positive families, the mutation/protein change is not specified,24,25 while in the remaining 6, all variants are located in the Ig repeats 1 to 7, within the Rod 1 domain (p.Gly288Arg, c.1829-1G>C, p.Pro637Gln, p.Val711Asp, p.Val761_Gln943del)8,22,23,29 (Figure 3). All these variants likely work as hypomorphic alleles by maintaining the reading frame of a presumably noncritical domain and thus may preserve some residual protein activity since they have been observed in hemizygous living males. The variant c.1829-1G>C was predicted to abolish the splicing acceptor site of intron 12, with a suggested subsequent activation of a cryptic acceptor site 15bp downstream in exon 13, which would result in an in-frame deletion of 5 consecutive residues (p.Phe611_Gly615del).22 Of note, a variant with the same predicted splicing effect was previously associated with PVNH and mild mitral valve dysfunction.18
The variant detected in our family was also predicted to destroy the canonical splicing acceptor site of intron 7, with the use of an alternative site located 24bp downstream in exon 8. In our case, the splicing alteration was confirmed by RNA studies, resulting in an in-frame deletion of 8 conserved amino acid residues (p.Val356_Gln363del). In-frame variants located in proximal Rod 1 domain of FLNA are considered to be associated with an isolated cardiac phenotype in affected male individuals. However, extracardiac findings in those patients suggest a rather continuous spectrum between partial and total loss of function variants (Table).
Cryptic acceptor sites, downstream of the canonical splice acceptor sites, are involved in the molecular mechanism of many human disorders. The use of these alternative sites to rescue the reading frame has been observed for other genes in which a partial loss of function may be better tolerated such as COL1A1 and COL1A2 in Ehlers-Danlos syndrome type VII vs osteogenesis imperfecta,30COL5A1 in Ehlers-Danlos syndrome types I and II,31DMD in Duchenne and Becker muscular dystrophies,32 and LAMA2 in autosomal recessive limb-girdle vs congenital muscular dystrophy.33 However, we cannot explain why patients with the 5-residue deletion in FLNA repeat 4 expressed such severe extracardiac manifestations, apparently observed even in their female ancestor, whereas the 4-exon deletion lacking repeats 5 to 7 and the present 8-residue deletion in repeat 1 are apparently restricted to an isolated cardiac phenotype. Different partner-dependent interactions in these regions, as well as other genetic factors, may be responsible for the observed phenotypes.
A role of skewed X-chromosome inactivation in the modulation of phenotypic expression in female carriers of FLNA pathogenic variants has also been proposed. However, an extremely skewed X-chromosome inactivation toward the mutated allele has only been persistently observed in FLNA-related skeletal dysplasias with congenital systemic involvement, which is consistent with a cell requirement of filamin A function during development.10 In loss of function alterations, however, no specific pattern has been described in association with the degree of disease manifestation.9 We observed a random X-chromosome inactivation pattern in 3 female carriers, all of whom were asymptomatic and one of them had mild, subclinical valve dysfunction. Thus, there seems to be no clinical correlation between phenotypic expression and X-chromosome inactivation pattern, supporting previous observations. Notwithstanding the foregoing, it should be noted that the X-chromosome inactivation process can be tissue-dependent and its determination in genomic DNA from lymphocytes may not be applicable to affected tissues.
Filamin A is known to bind several partner proteins besides actin filaments, thus probably explaining the reported diversity in the resulting phenotypes. Valvular phenotypic expression and possibly other soft tissue manifestations of FLNA-related disorders are thought to be associated with the interaction of filamin A with members of the TGF-β signaling pathway, which is already implicated in connective tissue disorders showing aortic root aneurysms and mitral valve prolapse.34 However, most of the interacting proteins identified to date do not bind to the N-terminal region of the Rod 1 domain, where mutations associated with XCVD have been described. Recently, a pathogenic role of these mutations has been functionally characterized as reducing cell spreading and migration capacities by means of an impaired Rho-GTPase signaling network modifying actin remodeling pathways that are important for cellular responses to mechanical stress, cell-extracellular matrix interactions, and epithelial-mesenchymal transformation.35
CONCLUSIONSOur findings support a role of in-frame variants in repeats 1-7 of the Rod 1 domain of filamin A in the expression of an XCVD main phenotype that can also include variably expressed connective tissue and possibly neurologic manifestations. An apparently isolated XCVD phenotype should prompt the search for these associated features beyond fibrillin and collagen disorders.36 Identifying genetic alterations in FLNA in these patients will confirm their diagnosis, enhance prognosis, and improve genetic counseling to their families.
FUNDINGThis work was partly supported by grant PI13/1450 from ISCII (Instituto de Salud Carlos III) and grant SAF2015-66831-R from MINECO (Ministerio de Economía, Industria y Competitividad), and confinanced by the FEDER (Fondo Europeo de Desarrollo Regional).
CONFLICTS OF INTERESTNone declared.
- –
X-linked cardiac valvular dysplasia is a rare congenital disorder that is predominantly expressed in males. It is caused by mutations in FLNA, the gene encoding membrane-cytoskeletal mediator protein filamin A. Unlike other autosomal forms of valvular disease, including connective tissue disorders, it can occur in an isolated manner, although some overlap among different filamin A-related phenotypes has been observed. It is thought that the different domains, mutation effects, interacting proteins, and genetic factors are involved in this variability.
- –
A novel mutation in FLNA has been found in a clinically described 3-generation family with XCVD. RNA studies in an affected male family member confirmed an in-frame deletion in the proximal Rod 1 domain and hence a partial loss of protein function as the predicted disease mechanism. Previously reported cases of XCVD have been reviewed, accounting for the predicted mutation location and protein damage when known, and extracardiac phenotypes when examined. A continuum of all filamin A loss of function-associated phenotypes is proposed, suggesting the need for their examination in patients with valvular heart disease.
The authors wish to thank all patients and relatives for their kind support.