Hypertrophic cardiomyopathy (HCM) is the most common inherited cardiac disease. The current challenge relies on the accurate classification of the pathogenicity of the variants. Transthoracic echocardiography (TTE) is recommended at initial evaluation and cardiac magnetic resonance (CMR) imaging should also be considered. We aimed to reappraise the penetrance and clinical expression of the MYBPC3 p.G263* variant.
MethodsThree hundred and eighty-four HCM probands and a control cohort of 450 individuals were studied for the main sarcomere genes by next-generation sequencing. All MYBPC3 p.G263* carriers were identified and family screening was performed. Clinical information was recorded retrospectively before 2015 and prospectively thereafter. Extra effort was invested in performing CMR in all carriers, despite TTE results.
ResultsThirteen HCM probands and none of the controls were carriers of the MYBPC3 p.G263* pathogenic variant (according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology). A total of 39 carriers were identified with family screening. Most patients with HCM were asymptomatic at the time of diagnosis and showed late-onset disease. Despite having a relatively benign course in the young, late HCM-related complications could occur. Penetrance was around 70% when evaluated by TTE and was 87.2% with TTE plus CMR. Penetrance was age-dependent, reaching 100% in carriers older than 55 years.
ConclusionsMYBPC3 p.G263* shares with most truncating pathogenic variants in this gene a late onset, relatively benign clinical course in the young, and high penetrance. Cardiac magnetic resonance could be a useful tool to evaluate carriers despite TTE results.
Keywords
Hypertrophic cardiomyopathy (HCM) has been defined as the presence of increased left ventricular wall thickness that is not solely explained by abnormal loading conditions.1 Hypertrophic cardiomyopathy is the most common inherited cardiac disease, affecting 1 in every 500 people, with a well-described pathological, clinical, and genetic profile.2 Transthoracic echocardiography (TTE) is recommended in all patients with HCM at initial evaluation (class I, level B), and cardiac magnetic resonance (CMR) imaging should be considered at baseline assessment if local resources and expertise permit.1
In most cases, HCM is inherited as an autosomal dominant genetic trait,3 with incomplete penetrance and variable clinical expression.1 Sequencing of sarcomere protein genes identifies a disease-causing variant in up to 60% of cases,3 with MYH7 (cardiac beta-myosin heavy chain) and MYBPC3 (cardiac myosin-binding protein C) accounting for most of the variants.4 Although genetic testing is recommended for all HCM patients, to enable genetic screening of their relatives (class I, level B), the lack of robust data on specific genotype-phenotype associations has reduced its impact on clinical management beyond screening.1 Due to the small number of families with specific pathogenic variants properly reported in the literature, conclusions about genotype-phenotype correlations are limited.5 Nevertheless, this situation should improve, as better data are being collected.1 Nowadays, the real challenge relies in the accurate classification of the pathogenicity of variants,2,6 where an extra effort should be invested. A deep knowledge of variants’ mechanism of pathogenicity in each gene is imperative to achieve a reliable classification. Apparently, most pathogenic variants in sarcomeric genes are missense and behave according to a dominant negative mechanism.5,7 However, in MYBPC3, most pathogenic variants seem to have a different mechanism of pathogenicity.8
Description of founder pathogenic variants provides a unique opportunity to assess clinical relationships between a specific genotype and a phenotype. We previously reported the novel MYBPC3 p.G263* variant, which was the most frequently reported variant in a region of northern Spain.9 The aim of this study was to reappraise the penetrance and clinical expression of this likely founder pathogenic variant.
METHODSStudy Population and Genetic AnalysisThree hundred and eighty-four index HCM patients from our referral unit for familial cardiomyopathies were studied for the main sarcomere genes by next-generation sequencing as reported by Gómez et al.10,11 In addition, we sequenced with the same gene panel 450 individuals of the RENASTUR healthy cohort, aged 60 to 85 years and without symptoms of HCM, as reported elsewhere.10–13 Genomic DNA of patients was isolated from blood samples. Written informed consent was obtained from each participant and the research protocol followed our institutional ethics guidelines.
Variant Classification AnalysisThe American College of Medical Genetics and Genomics/Association for Molecular Pathology (ACMG/AMP) recommends evaluating the pathogenicity of variants for Mendelian disorders with a probabilistic classification (“pathogenic”, “likely pathogenic”, “uncertain significance=VUS”, “likely benign”, and “benign”) based on multiple lines of evidence.6 Allele frequency > 5% in the Exome Sequencing Project, 1000 Genomes Project, or Exome Aggregation Consortium (ExAC) is used as stand-alone support for a benign interpretation. Thus, lower frequency variants reported since the 2015 ACMG/AMP guidelines, such as MYBPC3 p.G263*, were evaluated according to these documents.6
Clinical EvaluationMYBPC3 p.G263* carriers were identified and managed following HCM guidelines.1 A pedigree was drawn for each proband and all available relatives underwent genetic screening.
Clinical evaluation including family and personal history, physical examination, resting 12-lead electrocardiography, 24-hour Holter monitoring, TTE, and CMR if possible were performed. Clinical information was recorded retrospectively before 2015 and prospectively from 2015 to date.
Two patients died without CMR. An extra effort was required to perform CMR in all living MYBPC3 p.G263* carriers, regardless of previous TTE results. This was performed in everyone except in persons with claustrophobia, implantable cardioverter-defibrillator, pregnancy or with advanced age. A clinical diagnosis of HCM was made in the presence of increased left ventricular wall thickness (≥ 15mm or ≥ 13 in HCM first-degree relatives), measured by any imaging technique, that was not solely explained by abnormal loading conditions.1 The HCM sudden death risk calculator14 was used.
Statistical AnalysisWe used the SPSS v.19 statistical software package. Data are expressed as the mean ± standard deviation for continuous variables and as frequencies or percentages for categorical variables. The chi-square test or Fisher exact test was used to compare frequencies, whereas differences in continuous variables were evaluated with either the Student t test or Mann-Whitney U test. A P value < .05 was considered significant.
RESULTSNext-generation SequencingA pathogenic/likely pathogenic variant was identified in 34% of the HCM index cases (132/384), MYBPC3 being the most frequently affected gene (79/384 [21%]).
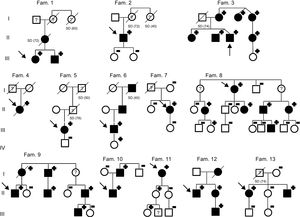
MYPBC3 p.G263* was identified in 13 apparently unrelated index cases (Figure 1) from the HCM cohort (13/384). The MYPBC3 p.G263* variant accounts for 10% of all pathogenic/likely pathogenic variants (13/132) found in this cohort and for 16.5% of those in the MYBPC3 gene (13/79). No other pathogenic/likely pathogenic variants were identified in these 13 patients (in our previous article, we reported the likely benign variant MYBPC3 R326Q, and the VUS MYH7 A100T).9

Pedigree of families with hypertrophic cardiomyopathy, carrying MYBPC3 p.G263*. Fam., family; SD, sudden death. Symbols denote sex and disease status: +, carriers; –, noncarriers; ?, unknown phenotype; box, male; circle, female; darkened, phenotype of hypertrophic cardiomyopathy; slashed, deceased; clear symbol, unaffected; without sign, not studied.
None of the 450 controls were carriers of MYBPC3 p.G263*.
Variant ClassificationAccording to ACMG/AMP,6MYBPC3 p.G263* is a pathogenic variant. It is a truncating variant in a gene in which loss of function is a known mechanism of disease, and computational evidence supports a deleterious effect on the gene. In addition, it is absent from controls in the Exome Sequencing Project, 1000 Genomes Project, and ExAC databases and in our RENASTUR control cohort. Furthermore, the robust segregation information in multiple affected family members (Figure 1) strongly supports its pathogenicity.
Study PopulationMYPBC3 p.G263* was identified in 13 index cases (mean age, 56 years ± 14.7 standard deviation, 6 men). A total of 66 individuals from these 13 families were evaluated (Figure 1). Family genetic testing identified 39 carriers (17 men) and 27 noncarriers.
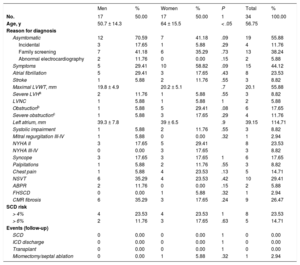
Thirty-four carriers (87.2%, 50% men) satisfied HCM criteria (Table 1). Disease penetrance, evaluated with TTE and CMR, was age-dependent, reaching 96.7% penetrance in carriers older than 40 years and 100% in those older than 55 years.
Characteristics of the 34 Affected Hypertrophic Cardiomyopathy Carriers of MYBPC3 p.G263X
| Men | % | Women | % | P | Total | % | |
|---|---|---|---|---|---|---|---|
| No. | 17 | 50.00 | 17 | 50.00 | 1 | 34 | 100.00 |
| Age, y | 50.7 ± 14.3 | 64 ± 15.5 | < .05 | 56.75 | |||
| Reason for diagnosis | |||||||
| Asymtomatic | 12 | 70.59 | 7 | 41.18 | .09 | 19 | 55.88 |
| Incidental | 3 | 17.65 | 1 | 5.88 | .29 | 4 | 11.76 |
| Family screening | 7 | 41.18 | 6 | 35.29 | .73 | 13 | 38.24 |
| Abnormal electrocardiography | 2 | 11.76 | 0 | 0.00 | .15 | 2 | 5.88 |
| Symptoms | 5 | 29.41 | 10 | 58.82 | .09 | 15 | 44.12 |
| Atrial fibrillation | 5 | 29.41 | 3 | 17.65 | .43 | 8 | 23.53 |
| Stroke | 1 | 5.88 | 2 | 11.76 | .55 | 3 | 8.82 |
| Maximal LVWT, mm | 19.8 ± 4.9 | 20.2 ± 5.1 | .7 | 20.1 | 55.88 | ||
| Severe LVHa | 2 | 11.76 | 1 | 5.88 | .55 | 3 | 8.82 |
| LVNC | 1 | 5.88 | 1 | 5.88 | 1 | 2 | 5.88 |
| Obstructionb | 1 | 5.88 | 5 | 29.41 | .08 | 6 | 17.65 |
| Severe obstructionc | 1 | 5.88 | 3 | 17.65 | .29 | 4 | 11.76 |
| Left atrium, mm | 39.3 ± 7.8 | 39 ± 6.5 | .9 | 39.15 | 114.71 | ||
| Systolic impairment | 1 | 5.88 | 2 | 11.76 | .55 | 3 | 8.82 |
| Mitral regurgitation III-IV | 1 | 5.88 | 0 | 0.00 | .32 | 1 | 2.94 |
| NYHA II | 3 | 17.65 | 5 | 29.41 | 8 | 23.53 | |
| NYHA III-IV | 0 | 0.00 | 3 | 17.65 | 3 | 8.82 | |
| Syncope | 3 | 17.65 | 3 | 17.65 | 1 | 6 | 17.65 |
| Palpitations | 1 | 5.88 | 2 | 11.76 | .55 | 3 | 8.82 |
| Chest pain | 1 | 5.88 | 4 | 23.53 | .13 | 5 | 14.71 |
| NSVT | 6 | 35.29 | 4 | 23.53 | .42 | 10 | 29.41 |
| ABPR | 2 | 11.76 | 0 | 0.00 | .15 | 2 | 5.88 |
| FHSCD | 0 | 0.00 | 1 | 5.88 | .32 | 1 | 2.94 |
| CMR fibrosis | 6 | 35.29 | 3 | 17.65 | .24 | 9 | 26.47 |
| SCD risk | |||||||
| > 4% | 4 | 23.53 | 4 | 23.53 | 1 | 8 | 23.53 |
| > 6% | 2 | 11.76 | 3 | 17.65 | .63 | 5 | 14.71 |
| Events (follow-up) | |||||||
| SCD | 0 | 0.00 | 0 | 0.00 | 1 | 0 | 0.00 |
| ICD discharge | 0 | 0.00 | 0 | 0.00 | 1 | 0 | 0.00 |
| Transplant | 0 | 0.00 | 0 | 0.00 | 1 | 0 | 0.00 |
| Miomectomy/septal ablation | 0 | 0.00 | 1 | 5.88 | .32 | 1 | 2.94 |
ABPR, abnormal blood pressure response during upright exercise; CMR, cardiac magnetic resonance; FHSCD, family history of sudden cardiac death; ICD, implantable cardioverter-defibrillator; LVH, left ventricular hypertrophy; LVNC, left ventricular noncompaction; LVWT, left ventricular wall thickness; NSVT, nonsustained ventricular tachycardia on Holter monitoring; NYHA, New York Heart Association; SCD, sudden cardiac death.
Unless otherwise indicated, data are expressed as No. (%) or mean ± standard deviation.
Of the 39 MYBPC3 p.G263* carriers, 24 underwent CMR, regardless of previous TTE results. Fibrosis was present in 37.5% of them. The CMR of 2 carriers with HCM exhibited features consistent with left ventricular noncompaction (LVNC). Cardiac magnetic resonance identified crypts unnoticed by TTE in 2 carriers without HCM.
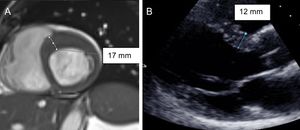
Cardiac magnetic resonance was the key to properly diagnosing 8 patients (Figure 2). Without CMR, left ventricular hypertrophy in 6 carriers would have gone unnoticed by TTE. Moreover, CMR helped assess a borderline hypertrophy identified in TTE and in a differential diagnosis between HCM and hypertensive cardiomyopathy. The average time between TTE and CMR was 1.2 years ± 1.25. Had CMR not been performed, penetrance rates evaluated with TTE only would have decreased to 66.7% to 71.8%. Cardiac magnetic resonance did not identify any false positive of TTE.
 and visualized on cardiac magnetic resonance (B).")
Men with the HCM phenotype were significantly younger than women. Interestingly, all clinically unaffected carriers were women younger than 55 years. These findings represent an overall 100% penetrance in men and 77.3% in women. Although there were other slight differences between sexes in clinical characteristics (Table 1), they did not reach statistical significance. The mean age at diagnosis was 50 ± 14 years in men and 59 years ± 15.5 in women. The mean age at genetic testing was 45 ± 15 years in male carriers, 54 ± 19 in female carriers, and 61 ± 15 in female carriers with HCM
Most patients (55.9%) were asymptomatic at diagnosis, especially in the context of family screening (Table 1). Although electrocardiography findings were not the main reason for cardiology referral, 85% of these findings in carriers were abnormal.
Only 1 patient had a family history of sudden cardiac death (SCD), when considered in first-degree relatives younger than 40 years (or any age if HCM was diagnosed). Nonetheless, SCD could appear at advanced ages and also in second-degree relatives (Figure 1).
The average SCD risk was low. However, in 9 patients the risk was higher (4 intermediate and 5 high). One patient with high risk died due to heart failure before implantable cardioverter-defibrillator implantation. An implantable cardioverter-defibrillator was implanted in the other 3 high-risk patients and implantation is pending in the fifth. Three of the 4 patients with intermediate risk also received an implantable cardioverter-defibrillator and implantation is being considered in the fourth. There were no cases of SCD or implantable cardioverter-defibrillator shocks. One patient's risk has diminished below 4% due to aging.
Only 3 patients developed left ventricular systolic impairment (being moderate/severe in the 2 with LVNC) and 3 had strokes (one with atrial fibrillation as a risk factor, another with LVCN and the third with both atrial fibrillation and LVNC).
DISCUSSIONAccurate Classification of VariantsThe growing public awareness of heritable diseases and the new available sequencing techniques have dramatically increased clinical demand for genetic testing, as well as the number of variants requiring bioinformatic and clinical interpretation.2 Nowadays, the number of HCM pathogenic variants reported is higher than expected. Walsh et al.15 analyzed data from 7855 individuals testing for inherited cardiomyopathies, along with 60706 ExAC reference samples. It was found that 11.7% of individuals in ExAC have reported HCM variants, an enormous excess of disease prevalence, incompatible with the actual prevalence of HCM (0.5%). This proved that many reported variants have been misclassified. For this reason, an extra effort should be invested in adequate classification of the pathogenicity of variants. Deep knowledge of the mechanisms of pathogenicity of each gene is essential and ACMG/AMP criteria6 should be analyzed with caution.
Cardiomyopathy-causing variants in most myofilament proteins incorporate into the sarcomere, acting as dominant negatives, such as missense variants in MYH7.5,7 In contrast, most pathogenic variants in MYBPC3 are truncating, causing HCM through haploinsufficiency.8,16,17 Walsh et al.15 confirmed the association between HCM and both the missense variation in MYH7 and the truncating variant in MYBPC3.16 They analyzed predicted truncating and nontruncating variants in cardiomyopathies and calculated their etiological fraction, which estimates the proportion of cases in which having a rare variant in a gene was disease-causing. In genes whose truncating alleles are disease-causing, the odds ratio, comparing rare-variant carriers with noncarriers, is typically higher. MYBPC3 nontruncanting variants had an odds ratio of 5.7 (0.8 etiological fraction) whereas truncating variants had an odds ratio that was nearly 21 times higher (0.99 etiological fraction). In contrast, MYH7 truncating variants had an odds ratio of only 1.7 (0.4 etiological fraction) and nontruncanting variants had an odds ratio of 12 (0.92 etiological fraction).15
Information about adequately characterized pathogenic variants should be reported to improve global databases and help their interpretation in other areas of the world where these variants may not be present.
Clinical Phenotype and Comparison With Other Pathogenic Variants in MYBPC3Although MYBPC3 pathogenic variants have been associated with a delayed onset of HCM (age > 40 years),18 mild hypertrophy, a low incidence of SCD, a relatively benign clinical course, and variability in the onset and prognosis of the disease has also been reported.19,20 Nevertheless, no differences in clinical phenotype have been attributable to a specific type of MYBPC3 variant.21 The high prevalence of founder pathogenic variants provides an opportunity to define their clinical profiles. Many founder pathogenic variants in MYBPC3 have been identified in some populations; interestingly, all of them were truncating variants17 and represented a large percentage (10%-25% to 58%) of the detected pathogenic variants in their countries of origin.22–29 The late onset of life-threatening HCM complications, delayed beyond reproductive age, has allowed its transmission to the next generations.24,25
Cardiac magnetic resonance enables 3-dimensional tomographic assessment of cardiac anatomy and has become an outstanding tool to evaluate cardiomyopathies. In fact, it is considered the noninvasive gold standard for the assessment of ventricular mass, volumes, and ejection fraction. Cardiac magnetic resonance is superior to TTE in the detection of left ventricular apical and anterolateral hypertrophy, aneurysms30 and thrombi.31 In some series, CMR has identified myocardial crypts in up to 70% of carriers of pathogenic variants without HCM.32–34 Although CMR is recommended in patients with inadequate echocardiographic windows (IB), sometimes its role in the HCM screening phase is limited, due to lack of availability of local resources.1 Thus, the penetrance of pathogenic variants in HCM is often evaluated based on TTE studies.
Unfortunately, in many studies of founder pathogenic variants, such as that by Adalsteinsdottir et al.,22 only probands or patients with known HCM phenotype were included, so disease penetrance could not be evaluated. The reported founder variant in MYBPC3 (c.927-2ANG transition in intron 11) is the main cause (58%) of HCM in Iceland.22MYBPC3 Gln1061X accounts for 17% of HCM cases in Finland26 and is associated with benign or intermediary phenotypes. Another Italian pathogenic founder variant, MYBPC3 p.F305Pfs*27, accounts for about 20% of HCM cases in their cohort.24 Patients carrying this variant also show late onset of HCM and SCD after the fourth decade of life.
The largest study performed to date, which compared CMR phenotypic characteristics among 125 HCM patients carrying MYH7 (53 patients) and MYBPC3 (75 patients) pathogenic variants, found no phenotypic differences between the 2 groups.35 Once again, unfortunately, this study was conducted in patients with already known HCM phenotype and differences in penetrance among patients harboring MYH7 and MYBPC3 pathogenic variants could not be assessed. However, the authors did find differences in patients older than 40 years: MYBPC3 carriers had significantly lower left ventricular ejection fraction.
In Japan, Kubo et al.,28 reported a 76.9% penetrance in their 39 MYBPC3 V592fs/8 carriers, although that study was based on TTE studies only (Table 2). Disease penetrance was 100% in participants older than 50 years and complications such as SCD and left ventricular systolic dysfunction with heart failure also occurred after the age of 50 years. MYBPC3 Val592fs/8 was identified in 16% of HCM probands.28
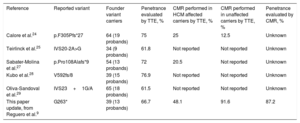
Comparison Between Founder Pathogenic Variants
| Reference | Reported variant | Founder variant carriers | Penetrance evaluated by TTE, % | CMR performed in HCM affected carriers by TTE, % | CMR performed in unaffected carriers by TTE, % | Penetrance evaluated by CMR, % |
|---|---|---|---|---|---|---|
| Calore et al.24 | p.F305Pfs*27 | 64 (19 probands) | 75 | 25 | 12.5 | Unknown |
| Teirlinck et al.25 | IVS20-2A>G | 34 (9 probands) | 61.8 | Not reported | Not reported | Unknown |
| Sabater-Molina et al.27 | p.Pro108Alafs*9 | 54 (13 probands) | 72 | 20.5 | Not reported | Unknown |
| Kubo et al.28 | V592fs/8 | 39 (15 probands) | 76.9 | Not reported | Not reported | Unknown |
| Oliva-Sandoval et al.29 | IVS23+1G/A | 65 (18 probands) | 61.5 | Not reported | Not reported | Unknown |
| This paper update, from Reguero et al.9 | G263* | 39 (13 probands) | 66.7 | 48.1 | 91.6 | 87.2 |
CMR, cardiac magnetic resonance; HCM, hypertrophic cardiomyopathy; TTE; transthoracic echocardiography.
Teirlinck et al.25 reported 61.8% HCM penetrance in 34 carriers of MYBPC3 IVS20-2A>G, based on TTE studies without CMR (Table 2). Interestingly, they were diagnosed later in life than carriers with other pathogenic variants in the other genes, despite having experienced the first symptoms at the same age. Presumably, the diagnostic delay was due to delayed appearance of ventricular hypertrophy.25
A study of another Spanish variant, based on TTE in 65 MYBPC3 IVS23+1G/A carriers, reported a penetrance of 61.5% (Table 2).29
Recent studies are beginning to include CMR in their analyses. A penetrance of 72% was reported in the novel Spanish variant MYBPC3 p.Pro108Alafs*9 (Table 2).27 However, in this cohort, CMR was only performed in 8 patients (14.8% of carriers).27
In the 64 carriers of the truncating variant MYBPC3 p.F305Pfs*2, disease penetrance was 75% (Table 2). Nevertheless, CMR was only performed in 12 affected carriers (25% of carriers) and 2 unaffected carriers.24
MYBPC3 p.G263* shares with other truncating pathogenic variants in MYBPC3 the high penetrance with low SCD risk profile and relatively benign clinical course, as previously described in the literature.9 This study provides a clinical update with 5 years’ follow-up, new information on relatives and diagnostic upgrades with CMR. In this pathogenic variant, late onset and life-threatening HCM complications also occurred, but at an advanced age, especially after 40 years, supporting the a priori benign profile of this pathogenic variant. In fact, many patients were asymptomatic at diagnosis (due to familial screening or as an incidental finding). A founder effect has not been proved, nor was it the objective of the study. However, both its high frequency (10% all pathogenic/likely pathogenic variants in HCM) and the historical and geographic isolation of our region, strongly supports the founder effect of the variant.
Not all HCM-related genes with pathogenic variants nor all pathogenic variants in the same gene behave the same way in terms of clinical presentation and outcomes.36 However, in general, pathogenic truncating variants in MYBPC3 seem to behave alike, despite the specific variant. Due to their late onset, their clinical course is, in general, benign. However, clinicians should be aware of possible late life-threatening complications (eg, SCD, stroke, left ventricular systolic impairment, and heart failure), particularly after the age of 40 to 50 years, in contrast to pathogenic variants in other HCM-related genes, whose “in-risk period” would have already been overcome.
Moreover, in our cohort, CMR helped to identify the HCM phenotype. Without CMR, penetrance rates would be similar to those reported in the other MYPBC3 variants (Table 2). In fact, the first time MYBPC3 p.G263* was reported in our population, an apparently low penetrance had been described.9 This reappraisal has allowed updating of its penetrance and clinical expression, identifying HCM patients who would otherwise have gone unnoticed. These data make us wonder whether, in fact, penetrance in other series may also have been underestimated, especially in young patients. As already suggested by Valente et al.,37 a percentage of individuals potentially affected by HCM may go unnoticed without CMR support. In their study, CMR identified mild hypertrophy in about 10% of pathogenic carriers with normal TTE wall thickness. As a result, in family screening of pathogenic variant carriers, when TTE is normal or when images are suboptimal, screening may be upgraded to CMR study.
In addition, in our population, CMR identified 2 patients in whom HMC coexisted with structural features consistent with LVNC. Therefore, this pathogenic variant represents an example of allelic heterogeneity, with variable phenotypic expression in carriers with distinct cardiomyopathies (HCM and LVNC), again raising the question of whether they are actually different manifestations of the same cardiomyopathy spectrum.38 The fact that the 2 patients in our population with LVNC had a stroke and moderate/severe systolic impairment, the 2 most severe complications of this cohort, makes this question intriguing.
LimitationsSome relatives declined to be clinically or/and genetically studied. Moreover, imaging analysis was not blind, since requests for CMR and TTE specified “HCM familial screening”. As the elapsed time between TTE and CMR tests was not homogeneous and was too long in some patients, caution should be exercised when interpreting these data. Finally, information about age-dependence penetrance is shown in this study. However, many affected patients were asymptomatic and were diagnosed by chance. Therefore, neither the age of first contact with a cardiologist was homogeneous nor could standardized imaging tests be performed (at the same age and with the same periodicity). Thus, assuming further conclusions about age-dependence data in this population was not possible.
CONCLUSIONSThe pathogenic variant in MYBPC3 p.G263* shares with most truncating pathogenic variants in this gene 3 characteristics: a late onset; a relatively benign clinical course in the young; and a high age-dependent penetrance. To identify all affected patients, CMR could be a useful tool to evaluate carriers despite an apparently normal TTE.
CONFLICTS OF INTERESTNone declared.
- –
The current challenge in HCM, the most common inherited cardiac diseases, lies in the accurate classification of the pathogenicity of variants.
- –
The lack of robust data on specific genotype-phenotype associations has reduced its impact on clinical management, beyond screening.
- –
We analyzed the pathogenic variant of MYBPC3 p.G263* and reviewed the literature on founder pathogenic variants, seeking possible genotype-phenotype correlations. This variant shares with most truncating pathogenic variants in this gene, the late onset and relatively benign clinical course in the young and a high age-dependent penetrance. Moreover, penetrance could be even higher if evaluated by CMR.