Keywords
The 21st century represents the dawn of a new era, an era in which most of the disease-causing genes will be discovered. The challenging task at hand will be to determine the function of these genes and their regulation in normal and disease states. Cardiology, like most of the other disciplines of medicine, has made significant advances in the field of molecular genetics. Today many genes associated with cardiac diseases have been identified (as shown in Table 1) and are being studied aggressively in laboratories around the world.

The human heart responds to physiological and pathological stimuli such as hypertension or valvular diseases by developing hypertrophy, dilatation, or a combination of these two responses. Thus, understanding the molecular basis of these phenotypes will not only provide significant insight into the pathogenesis of inherited diseases but also into acquired diseases that give rise to these common phenotypic features. It is believed that in hypertrophy the sarcomeres are laid down in parallel with thickening of the wall and that in dilatation the sarcomeres are added in sequence with a normal or thinning wall. It will be interesting to understand the mechanisms that direct the heart towards hypertrophy versus dilatation. Despite the limited phenotypic response, the signaling pathways are numerous and complex. It is here that familial or inherited forms of cardiomyopathy, i.e., FHCM (familial hypertrophic cardiomyopathy) and FDCM (familial dilated cardiomyopathy) provide a surrogate for both these phenotypic end-points and hence serve as excellent models for studying the growth response of the heart to both physiologic and pathologic stress. It is important to stress that although both FHCM and FDCM are monogenic disorders, the final phenotype results not only from the primary causal mutations but also their interactions with other genes and surrounding environmental stimuli.
HYPERTROPHIC CARDIOMYOPATHY
HCM is characterized by hypertrophy of the left ventricle in the absence of increased afterload. Usually the LV cavity is small, with preserved or increased LVEF, and later the stiff ventricle gives rise to impaired relaxation resulting in diastolic dysfunction which is responsible for most of the signs and symptoms of heart failure. The clinical course ranges from asymptomatic to severe CHF, and includes syncope and SCD. The latter is often the first presentation, and HCM is the leading cause of SCD in young athletes.1,2 The pathologic phenotype of HCM consists of cardiac myocyte hypertrophy, disarray, increased interstitial collagen deposition, and intimal hypertrophy of intramural coronary arteries. Myocyte disarray is considered the pathologic hallmark of HCM.3,4 The prevalence of HCM as diagnosed by echocardiography is estimated to be 1:1000 in young individuals5 and is thought to increase with age.6 This age-associated increase may reflect the age-dependent penetrance of certain mutations, e.g., as observed with myosin-binding protein C mutations (MBPC).7
GENETICS OF HCM
At present, HCM is considered to be a disease of the sarcomere because all the genes responsible for HCM identified so far encode for sarcomeric proteins. HCM is inherited in an autosomal dominant mode. Each individual has two copies of each gene, called alleles. In the case of autosomal dominant disease, a single defective allele is enough to cause the disease, whereas in the case of autosomal recessive disease, both alleles have to be defective in order to cause disease. HCM exhibits genetic heterogeneity, with multiple loci (8 genes) and each gene having multiple mutations, as seen in Table 2.

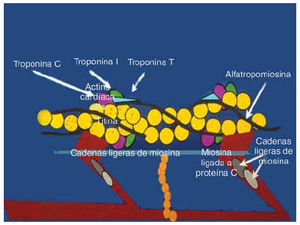
The familial nature of a disease is determined from the family history. In the era prior to molecular genetics the disease was considered to be of familial nature only when the family history was available. Cases that did not have such a family history were considered sporadic. It has been proven in the case of HCM that these sporadic cases are of genetic nature as they occur due to mutations arising de novo in the sarcomeric genes and subsequent transmission to the offspring occurs in an autosomal dominant mode.8 Beta-MCHC was the first gene identified to cause HCM.9 Most of the mutations found in β-MCHC are missense mutations that occur mainly in the region encoding the globular head region of the protein, which interacts with the actin filament. The second most common gene causing HCM is the myosin-binding protein C (MBPC)10, which binds to thick filaments in the region of A-bands and titin, the latter being a large sarcomeric protein that stretches from one Z-band to the other. Most of the mutations in MBPC are deletions that result in frameshifts or premature truncation and affect protein-binding sites. The mutations in cardiac troponin T (cTnT) are mostly missense and account for about 15% of HCM.11 This protein is a major component of the troponin complex, which has an important role in cardiac contractile function by regulating calcium homeostasis.12 Codon 92 is considered a hot spot for these mutations.13 Alpha-tropomyosin,14 cardiac troponin I,15 and the essential regulatory light-chain proteins MLC1 and MLC216 are less common causes of HCM. Briefly, α-tropomyosin is responsible for the positioning of the troponin complex on the actin filament and the light chains bind to the α-helices of the β-MCHC protein. Cardiac troponin I is very specific for cardiac tissue and forms the inhibitory component of the troponin complex, modulating the acto-myosin stimulated ATPase activity. Recently, cardiac actin has been identified as a cause of HCM.17 This is of particular interest, as cardiac actin has also been known to cause DCM. So far, it is the only gene linked to both phenotypes. Actin is a sarcomeric protein that interacts with the cytoskeletal proteins by anchoring to actinin and dystrophin.
Genotype - phenotype correlation
To some extent there has been a limitation in establishing the genotype-phenotype correlation due to the small size of the families, variable penetrance and expressivity, low frequency of each mutation, and the effect of non-genetic factors. However, a consensus has been established for some of the genes and the prognostic significance of their mutations determined primarily for the β-MHC mutations. In the case of β-MHC, some correlations have been established with the degree of hypertrophy, the penetrance, and the incidence of SCD.18-20 The β-MHC mutations Arg403Gln, Arg719Trp, and Arg453Cys are considered malignant as they are associated with a high incidence of SCD, severe hypertrophy, and high penetrance. The average life expectancy in these mutations is 30-38 years. In contrast, the mutations Val606Met and Leu908Val are associated with mild hypertrophy, a benign course, and almost normal life expectancy, whereas the β-MHC mutations Glu930lys and Arg349Gln are of intermediate severity, with a life span of about 45 years. The prognostic significance of mutations in β-MHC correlates with the degree of hypertrophy, penetrance, and a high incidence of SCD.21 The mutations of myosin-binding protein C (MBPC) gene are mostly small deletions and are associated with late age of onset (4th5th decade), mild hypertrophy, and a relatively benign course with near normal life expectancy. Mutations in cTnT are associated with mild or almost no hypertrophy but a high incidence of SCD, especially in young men.22 This suggests, at least for this gene, that there is a dissociation between the degree of hypertrophy and the incidence of SCD. There is, however, increased fibrous tissue and sarcomere disarray, which is also observed in genetic animal models (Table 3). It is believed that other genetic factors may also influence the extent of hypertrophy, e.g., variants of the ACE gene are known to influence hypertrophy.23 Alpha-tropomyosin mutations are mostly missense and are associated with mild hypertrophy, low penetrance, and a benign course. Recently, a missense mutation in this gene was described, which is associated with a more malignant phenotype of mild hypertrophy but high incidence of SCD similar to the troponin T mutations.24

In summary, the important observations from genotype-phenotype studies are: firstly, that clinical features do not reliably predict SCD; secondly, the disease seldom develops until puberty or later, and thirdly, certain mutations are predictive of SCD.
Pathogenesis of HCM
The results of several in vitro and in vivo studies have provided a framework for the pathogenesis of familial hypertrophic cardiomyopathy (FHCM)25-26. The following observations have played a pivotal role in our present understanding:
1. In vitro studies with several β-MHC and troponin T mutations indicate that the defect impairs some aspect of contractility through mechanisms such as decreased binding of myosin to actin or decreased generation of ATPase activity.
2. In vitro functional assessment of cardiac or skeletal muscle fibers from patients with β-MHC mutations exhibit decreased contractile activity.
3. Functional assessment of β-MHC of troponin T mutations responsible for FHCM expressed in adult feline myocytes show impaired contractility.
4. In vitro studies in the feline myocyte suggest that impaired contractility precedes sarcomere disarray, which was confirmed in a transgenic mouse.
5. Studies in multiple in vitro and in vivo preparations show that the mutant protein is incorporated into the sarcomeric myofibrils.
6. Genetic animal models (transgenic and knockout) expressing β-MHC and troponin T mutations have confirmed a dominant-negative mechanism to be responsible for the phenotype, in keeping with the «poison peptide» hypothesis.
7. The hypertrophy and increased fibrous tissue appear to be compensatory responses to the genetic defect, presumably due to release of growth factors.
Mutant protein is incorporated into the sarcomere and causes contractile dysfunction by various mechanisms
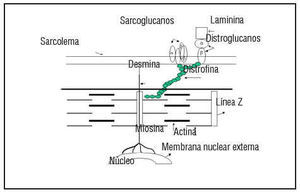
The fact that all of the genes associated with HCM encode for sarcomeric proteins defines HCM as a disease of the sarcomere (Figure 1). The sarcomere forms the basic unit of contraction in cardiac muscle and, therefore, it was reasonable to postulate that the basic defect is contractile dysfunction.27 The first studies done have shown that the mutant protein is, in fact, incorporated into the sarcomere.28-30 Evidence of the contractile dysfunction of the sarcomere secondary to the mutation comes from the study of single-cell preparations. Studies made with both skeletal and cardiac muscle fibers expressing a β-MHC mutation have shown decreased mechanical performance.31-34 Marian et al29 showed that incorporation of β-MHC mutant protein in the adult feline myocyte causes contractile dysfunction by decreasing cell shortening. Similarly Rust et al30 showed that the adult cardiac myocyte expressing mutant cTnT proteins shows contractile dysfunction by exhibiting significant desensitization to activation by calcium. The mutations in the sarcomeric proteins cause contractile dysfunction by affecting its various functions. The ability of the mutant β-MHC proteins to dislocate actin filaments has been assessed using an in vitro motility assay, in which the β-MHC molecules are fixed to a membrane.32,35-37 This could be due to reduced binding to actin as evidenced by an increased rate of dissociation.37 The mutations in myosin have been shown to decrease the actin-activated ATPase activity. This may reflect the decreased affinity of the myosin filament for the actin filaments.37 Also the calcium sensitivity of the sarcomeric proteins may be altered, affecting the acto-myosin interaction.30 Two of the mutations in the α-tropomyosin gene have been shown to alter the calcium-dependent38 acto-myosin interaction. As this research was evolving from molecular genetics, Rayment et al39 published the three-dimensional structure of beta-myosin heavy-chain protein from skeletal muscle. All of the mutations of the β-MHC can be superimposed on the functional domains and, as expected from the functional studies, have been shown to affect the functions associated with these domains, such as actin-binding, ATPase generation, and calcium sensitivity. Thus, these data indicate that affectation of various functional aspects of its interaction impairs cardiac myocyte function following expression of mutant sarcomeric proteins.

Fig. 1. All genes identified causing FHCM encode for the sarcomere proteins. The schematic detail of the structure of sarcomere is shown here.
Studies performed with skeletal and cardiac muscle fibers expressing a β-MHC mutation were shown to have decreased mechanical performance.31-34 Studies performed in the intact feline myocyte showed that mutations of β-MHC were associated with sarcomere disarray.29 Similarly, expression of mutant cTnT proteins in the feline cardiac myocyte30,40 and myotubes41,42 show sarcomere disarray. Expression of the mutant cTnT-Gln92, known to cause HCM in man,22 in adult feline cardiac myocytes was associated with decreased cell shortening. Impaired contractile performance was also observed in rat cardiac myocytes.30 This effect was dominant-negative and preceded the development of sarcomere disarray in the myocytes.40 The shortening velocity of myotubes42 was observed to be decreased upon expressing the mutant cTnT-Gln92 protein. Myocytes isolated from the heart of an α-MyHC403 heterozygote mouse exhibited impaired contraction and relaxation.43 Thus, these data indicate that cardiac myocyte function following expression of mutant sarcomeric proteins is impaired and strongly suggest that impaired contractility preceded sarcomere disarray. It is noteworthy that in the feline myocyte preparation the mutant protein was shown to be incorporated into the sarcomeric myofibrils.
Effect of mutation on assembly of filaments
The potential of the mutant protein to affect the assembly of the filaments in the sarcomere is being explored. Recently Muraishi et al44 showed that in cases of human HCM mutations in the β-MHC cause the alignment of the sarcomeric filaments to be disrupted. The distance between the neighboring myosin filaments was increased and the sarcomeric filament alignment was also sparse and disrupted. Chien et al28 had shown in cultured neonatal rat ventricular cardiomyocytes that while mutations in the ATP-binding site of β-MHC disrupt the filament assembly, the β-MHC mutations associated with familial HCM did not do so, suggesting that the myocyte disarray may be secondary to contractile dysfunction resulting from the mutation. The type of mutation may be important with the regard to its ability to cause disruption of the sarcomere assembly.
Functional changes precede morphological changes. Evidence for the compensatory nature of the hypertrophy
Considerable evidence now shows that left ventricular dysfunction precedes the morphological changes observed.30,30,45 In vitro studies in adult feline cardiac myocyte by Marian et al29 provided the first clue that the expression of the mutant sarcomeric protein impaired contractile dysfunction prior to any discernable sarcomere or myofibrillar disarray. Recently published data by Kass et al46 of the in vivo study of pressure-volume in transgenic mice with the alpha-myosin chain mutation Arg403Gln indicate that young mutant mice (6 weeks) show altered contraction kinetics with delayed pressure r elaxation and chamber filling. Older mutant mice (20 weeks) showed diastolic and systolic changes with hyperdynamic contraction, increased end-systolic stiffness, and decreased cardiac index. This provides additional confirmation as the young mice previously were shown not to exhibit any histologic abnormalities at 6 weeks of age and adult mice with the disease had shown myocyte disarray and fibrosis. T he contractile dysfunction serves as a stimulus for growth factors, which then lead to cardiac myocyte hypertrophy and increased interstitial collagen. This, in turn, gives rise to the hypertrophy phenotype observed. Thus, the hypertrophy observed is compensatory, occurring secondary to contractile dysfunction of the cardiac myocyte. It is an interesting fact that hypertrophy occurs only in the left ventricle despite expression of mutant β-MHC protein in the right (low-pressure chamber) and left (high-pressure chamber) ventricles. Molecular markers of hypertrophy, such as TGF beta and IGF-1, expressed in response to an increased load are also expressed in the hypertrophy of FHCM (Li et al., 5060) suggesting that common downstream pathways may be involved in the hypertrophic response to both acquired and genetic stimuli. Also of note is the absence of significant clinical abnormality in the skeletal muscles of patients with HCM, despite expression of the mutant β-MHC All of the above evidence suggests that the activation of the genetic program that leads to hypertrophy must involve multiple genes, which would be modulated by a variety of genetic and non-genetic factors such as local environmental milieu.
«Poison peptide» theory
It is now the prevailing hypothesis that the mutant sarcomeric protein is incorporated into the myofibril and acts as a «poison peptide». This then leads to contractile dysfunction of the sarcomeric unit. This results in an impaired rate of cell shortening, as detected by laser and seen in the expression of the mutant cTnT protein in the mouse model. Also in this cTntGln92 model, Marian et al showed that the increased expression of the mutant protein was associated with a more severe phenotype, confirming the dominant negative nature of the mutant protein.45
Observations in transgenic models
The results of expressing β-MHC and troponin T mutations as transgenes in the mouse have confirmed sarcomere disarray is a fundamental hallmark of the phenotype. The morphological and functional changes observed in the various genetic animal models are summarized in Table 3.47-55 Sarcomeric disarray and increased fibrous tissue have been consistently observed in all animal models with expression of the mutant protein, while absent with the expression of the wild type protein (normal). Furthermore, these studies have confirmed that the mutant is dominant-negative, in keeping with the poison peptide hypothesis.56,57 It noteworthy, however, that in the mouse model hypertrophy is mild in the heterozygotes with essentially all of the mutations expressed. The mechanism responsible for sarcomere disarray and increased fibrous tissue but minimal hypertrophy in the mouse remains unknown. Recently, a transgenic rabbit model expressing the b-MHC (Arg403Gln) mutation was developed and was shown to exhibit virtually the same morphologic changes as in adult humans, including sarcomeric disarray, increased collagen, and hypertrophy.55 The rabbit model has several advantages over the mouse, namely a) its native endogenous myosin is β-MHC, as in humans; b) the heart is much larger; c) the electrophysiology of the rabbit heart has been well characterized. These features provide the opportunity to do serial noninvasive testing in a model in which β-MHC is endogenous. The well-characterized rabbit model for sudden death provides important baseline data for comparisons to the newly induced phenotype.
In summary, the mutant sarcomeric protein is incorporated into the myofibril and acts as a «poison peptide,» resulting in contractile dysfunction of the myocytes. This dysfunction triggers the release of growth factors, which then leads to the final phenotype of hypertrophy, myocyte disarray and fibrosis. This final phenotype is also modified by the interplay of primary mutation with other genetic factors and the environmental milieu.
Clinical implications
Due to the significant variability in phenotypic presentation, age of onset, and the course of disease associated with genetic heterogeneity, it is difficult to suggest a common guideline which would be applicable in clinical practice for counseling. The diagnosis is further confounded in the presence of hypertension or valvular disease.
In spite of these limitations, genetic testing would provide important information in families with a history of HCM. FHCM is an autosomal dominant disease, with offspring of the affected individual having a 50% chance of getting the disease. Genetic testing would identify the 50% who are at risk of developing the disease and, depending on the type of mutation, help in assessing the prognosis. In younger family members who do not have clinical or echocardiographic evidence of hypertrophy, it would be an invaluable tool for determining those at risk and would provide a window of opportunity to apply treatment or to prevent the phenotype from developing. Furthermore, genetic testing will help to develop the necessary database that will ultimately be used to routinely screen for mutations. At the present time, another limitation is the unavailability of a genetic test that is not time-consuming and expensive. However, with the advent of DNA microchip-array technology, it will be easy to screen even sporadic cases of HCM for known mutations in a cost-effective manner. The different mutations have different phenotypic characteristics and it also is of great significance that SCD may not be associated with significant hypertrophy in some of these mutations. However, it cannot be emphasized enough that as HCM usually presents after puberty, there is a tremendous window of opportunity for therapeutic interventions in the future to prevent the disease in individuals with the genetic defect if diagnosed early. In fact, preventing sudden death from ischemic cardiac disease must now be entertained, at least in symptomatic individuals.
DILATED CARDIOMYOPATHY
Heart failure is the most common manifestation of DCM, a leading cause of cardiovascular mortality. Idiopathic DCM accounts for about 30% of cases, with manifestations secondary to ischemic and infectious causes accounting for the remaining 70%. Idiopathic DCM, by definition, is the presence of left ventricular dysfunction with decreased ejection fraction and left ventricular dilatation in the absence of any known cause. Left ventricular ejection fraction of <45% and left ventricular end-diastolic diameter of >2.7cm/m² are the recommended criteria for the diagnosis of DCM (WHO/ISFC Task Force, 1980). The signs and symptoms of heart failure are the leading mode of presentation and others include cardiac arrhythmias, SCD, and thromboembolism. The prevalence of idiopathic DCM is estimated to be 40 cases per 100 00058, with familial DCM accounting for about 30%.59 Genetic causes probably account for an even larger number of cases of DCM but these are sporadic due to de novo mutation and not familial.
Genetic DCM can be categorized based on the mode of inheritance into autosomal, X-linked DCM, and mitochondrial DCM. The familial DCM of autosomal dominant inheritance can be further classified as DCM alone and DCM with cardiac conduction disturbances. The loci for DCM without conduction disorders have been mapped to 1q32, 9q13, 10q24 and 2q14. The loci for DCM with conduction disturbances are 1p1-1q1, 3p22-25, 6q23 and 2q31. The conduction disturbances include second or third-degree AV block, atrial fibrillation, or marked bradycardia. DCM usually develops by the fourth or fifth decade and sudden cardiac death occurs commonly in late stages of the disease. So far, none of the genes responsible for DCM at these loci have been identified.
The cardiac actin gene was recently found to have missense mutations which cosegregated with DCM in two unrelated families.60 Thus, cardiac actin became the first gene to be known to cause hereditary DCM. In both of these families, mutations were identified in domains of actin that attach to the Z-band and intercalated disks and involve universally conserved amino acids. The cardiac actin gene was shown recently to cause familial HCM. However the mutation responsible for HCM occurs in the domain affecting the myosin crossbridge. This is of significant importance and will be addressed later under «Paradigm of DCM.»
Recently, our laboratory identified desmin mutations in a family with DCM.61 Desmin is a large cytoskeletal protein, which stretches from the Z-band in the sarcomere to the nuclear membrane and other organelles in the cell, and is known to impart mechanical stability to the cells.62 A missense mutation was identified in our family, which has DCM without any skeletal or smooth muscle abnormalities. Desmin has been previously shown to be associated with skeletal and cardiac abnormalities. The mutations in these families with skeletal and cardiac combined abnormalities have been in the rod region of the protein. In our family with a cardiac-restricted phenotype, the mutation was found in the tail region of desmin, which would suggest a possible unique cardiac function of this domain. This is of some functional significance because while several functions have been ascribed to the rod region, the function of the tail region remains controversial.
X-linked DCM
In 1987, Berko and Swift63 reported a five-generation history of DCM and no skeletal myopathy evident. The males presented in their teens or early twenties with evidence of mitral regurgitation and echocardiographic evidence of DCM. The disease progressed fast, leading to death or cardiac transplantation within 1-2 years. The females who manifested the disease did so in the fourth or fifth decade and the disease progressed slowly. The postmortem study showed marked dilatation with widespread patchy fibrosis and normal mitochondria on electron microscopy. There was no difference in pathognomic findings compared to other idiopathic DCM except that the inheritance was X-linked.
Ortiz-López et al64 showed the linkage of X-linked DCM to the dystrophin locus on Xp21 (gene known to responsible for Duchenne and Becker muscular dystrophy) in these families. Evaluation of the protein showed absewas normal. Also dystrophin-associated glycoprotein (α-dystroglycan) was decreased in the cardiac tissue. Dystrophin is one of the largest known proteins and has four domains: an actin-binding site, triple helical central repeat, a cysteine-rich region, and a carboxy-terminal region. In the cardiac-specific phenotype, the level of this protein is reduced only in the cardiac tissue.
In one case of DCM, absence of metavinculin was reported.65 Metavinculin is an alternatively spliced form of vinculin,66 and is part of a cytoskeletal protein localized in the intercalated disk. The function of metavinculin is unclear and whether, indeed, it causes DCM remains to be proven.
Current paradigm of DCM
Since most of the genes responsible for familial DCM have not been identified, it would be premature to speculate on the mechanism for the pathogenesis of the disease. The genes identified to date to cause DCM, namely dystrophin and desmin, encode for cytoskeletal proteins and cardiac actin, though a sarcomeric protein is related to the cytoskeleton since it anchors to actinin and dystrophin (Figure 2). Also, the actin mutation that occurs in association with HCM occurs in the region that encodes a domain that binds to myosin whereas the mutations responsible for DCM occur in a domain that anchors to the Z-band and intercalated disks. This gives rise to the interesting hypothesis that a mutation in the myosin-binding domain of actin gives rise to sarcomeric contractile dysfunction and hence results in HCM, whereas the mutation in the domain anchoring to intercalated disk and Z-band gives rise to an abnormal force of transmission which results in DCM.

Fig. 2. Schematic diagram showing the relation of the sarcomeric proteins to the cytoskeleton. Cardiac actin, a sarcomeric protein, anchors to dystrophin and actinin, cytoskeletal proteins. The mutation in cardiac actin is known to cause both HCM and DCM, depending on which domain it occurs in. Dystrophin and desmin are the only two cytoskeletal proteins known to cause human DCM . d-sarcoglycan is known to cause DCM in an animal (hamster) model of the disease.
There is evidence that other cytoskeletal proteins may also be involved. The BIO 14.6 hamster, a model for human hereditary cardiomyopathy,67 showed a common gene defect in (-sarcoglycan, which resulted in deficiency of δ-SG transcription and consequent loss of δ-SG protein with resultant HCM and DCM. In these models an interesting observation was made: the mice exhibited substantial loss of sarcoglycan complex in the vascular smooth muscle, which was associated with vascular malfunction and irregularities of coronary arteries. This gives rise to an interesting hypothesis that vascular defects may play an important role in the development of cardiomyopathy and may be an avenue that can be targeted for therapeutic intervention. There is further evidence from recent studies to support the central role played by the cytoskeletal proteins in dilatation of the heart. Mice lacking a muscle-specific LIM domain protein, MLP, display a severe form of dilated cardiomyopathy with several features similar to the human disease.68 MLP is localized in the actin-based cytoskeleton in the myocyte. MLP contains two LIM domains, which serve as a module for protein-to-protein interactions, which link MLP to other cytoskeletal proteins.
It is now believed that the defect in the cytoskeletal proteins serves to trigger a cascade of events that ultimately result in the dilated cardiomyopathic phenotype. The trigger may be in the form of either defective transmission of force of contraction or loss of sarcolemmal integrity. The cascade of events triggered is just now being elucidated. The recent discovery of myocyte survival pathways has provided the strongest evidence to date that the loss of functional myocytes may trigger the onset of dilation of the chamber in response to biomechanical stress. Cardiotropin 1 (CT-1), a recently identified member of the IL-6 cytokine family, is believed to be one of most potent cardiac myocyte survival factors,69 which in part acts through the gp-130 pathway and blocks myocyte apoptosis.70 Apoptosis is programmed cell death that occurs without any inflammatory changes. There is emerging evidence that apoptosis exists in dilated cardiomyopathy and is responsible for loss of cardiomyocytes with loss of cardiac mass. The loss of cardiac mass may then lead to pump failure and also cause electrical conduction consistent with resultant arrhythmias. Mice that have ventricular restricted knockout of gp-130 exhibit a normal basal cardiac phenotype but display a rapid onset of dilatation in response to biomechanical stress. It is known that biomechanical stress activates a compensatory hypertrophic response normally and the above study also showed that it does activate a proapoptotic cascade, which is not expressed normally in the presence of the CT-1-gp-130 pathway. This hypothesis proposes that biomechanical stress activates both the hypertrophic cascade and proapoptotic programs concomitantly, but the proapoptotic pathway is held in check by CT-1 and hypertrophy is the compensatory response. In contrast, the transition to cardiac failure occurs via constitutive activation of the proapoptotic pathway. What causes the genetic program to switch from hypertrophy to an apoptotic response? Why does it lead to chamber dilatation? Why in DCM is the apoptotic program expressed preferentially? Adams et al have shown that moderate levels of Galfaq signaling stimulate cardiac hypertrophy in cultured neonatal cardiac myocytes. The Galfaq family of proteins are signal transducers for a variety of tissue-specific effects. Cardiac muscle expresses Galfaq-coupled receptors that are now believed to mediate hypertrophy. In transgenic mice overexpressing Galfaq, compensated hypertrophy rapidly progressed to lethal cardiomyopathy as a consequence of severe apoptosis. These data suggested that moderate levels of Galfaq cause hypertrophy whereas high levels of Galfaq cause cardiomyocyte apoptosis. Thus, a single biochemical stimulus regulating cardiomyocyte growth and programmed cell death at different levels of expression is an intriguing mechanism by which compensated hypertrophy may progress to decompensated heart failure.71 The factors that may influence the level of expression of Galfaq have yet to be elucidated. Again, it will be interesting to find out whether in DCM levels of Galfaq expression are high to begin with and what triggers this response.
There is also evidence of the role played by immune factors in contributing to the phenotype of DCM. Recently, MCP-1 (monocyte chemoattractant protein) messenger mRNA was detected in the endomyocardial biopsy tissue of patients with DCM. Furthermore, a study by Lehmann et al72 showed that the severity of LV dysfunction is related to the levels of expression of the MCP-1 messenger RNA. MCP-1 regulates leukocyte migration and tissue infiltration, which may lead to myocyte damage.
Recently, Badorff et al73 showed that enteroviral protease 2A cleaved dystrophin in in vitro studies, which resulted in impaired dystrophin function and increased cell permeability. This presents a very intriguing hypothesis that even acquired DCM may share the same final common pathway as familial DCM.
Current evidence suggests that a chronic increase in wall stress mediated via the cytoskeletal proteins and related path ways may lead to concomitant activation of the hypertrophic, apoptotic myocyte survival programs that lead to the final phenotype of dilated cardiomyopathy and heart failure. Thus, interruption of these pathways at the molecular level will be targets for the development of specific drugs to either arrest or reverse the process of dilated cardiomyopathy.
ARRYTHMOGENIC RIGHT VENTRICULAR DYSPLASIA (ARVD)
ARVD is characterized by fatty infiltration of the right ventricle, fibrosis, and finally thinning of the wall with chamber dilatation. This is a difficult disease to diagnose and at times to distinguish from DCM. It is the most common cause of SCD in young athletes in Italy.74 It is also said to account for about 17% of SCD in young athletes in North America.75
Three loci with an autosomal dominant mode of inheritance were mapped in an Italian population, 1q42-43, 2q32, and 14q12, and a fourth locus, autosomal recessive, associated with Naxos disease was mapped in a Greek family to 17q. Recently, in North America two loci responsible for ARVD have been mapped at 3p23 and 10p12. None of the genes in these loci, causative of the disease, have been identified.
This extremely difficult disease to diagnose affects only the right ventricle, initiating in the epicardium and spreading to the endocardium. Thus, right ventricular biopsy is definitive when positive but a negative biopsy does not rule out the disease, as the lesion may be inaccessible. Consensus diagnostic criteria have been developed utilizing right ventricular biopsy, MRI, echocardiography, and electrocardiography. The observed EKG abnormalities include inverted T waves in the right precordial leads, late potentials, and right ventricular arrhythmia with LBBB. Often the first presentation is SCD. Not much is known or understood regarding the pathogenesis of this disease. Its predominant affectation of the right ventricle with fatty infiltration remains a mystery. Recently, evidence has been found suggesting that apoptosis may play a role in this disease process. The discovery of a gene causing the disease would be a major step forward in making the diagnosis and elucidating the pathogenesis of this disease.
ACKNOWLEDGEMENT
We greatly appreciate the secretarial assistance of Debora Weaver and Valorie Garza in the preparation of this manuscript and figures. This work is supported in part by grants from the National Heart, Lung, and Blood Institute, Specialized Centers of Research (P50-HL42267-01), and the American Heart Association, Bugher Foundation Center for Molecular Biology (86-2216).
Correspondencia: Dr. R. Roberts.
Section of Cardiology. Baylor College of Medicine.
6550 Fannin St SM 677. Houston, Texas 77030. USA.
E-mail: rroberts@bcm.tmc.edu