
Keywords
INTRODUCTION
Hypertrophic cardiomyopathy (HCM) is the most common reason for sudden cardiac death in young adults, and a major cause of morbidity and mortality in the elderly. The associated clinical picture is highly variable and ranges from incapacitating symptoms to no symptoms at all. Many patients remain asymptomatic for long periods, although the percentage of patients with severe symptoms increases with age.1
It has been estimated that 0.2% of individuals have a wall thickness ≥15 mm.2 The pathophysiological basis of HCM is mutation of genes encoding the sarcomeric proteins. Around 30% to 40% of patients are diagnosed as sporadic cases, although the incomplete penetration of some mutations could mean that the percentage of true familial cases is underestimated.3 The clinical heterogeneity is primarily due to the fact that at least 12 genes can mutate. The first mutations were found in the MYH7gene, which encodes cardiac beta myosin heavy chain.4-7 Mutations were later identified in other genes, such as TNNT2 (troponin-T) and MYBPC3(cardiac myosin-binding protein C).8-14
Most of the mutations have been found in a single family, which makes it difficult to obtain conclusive data on the phenotype associated with each mutation. Relevant data on the genotype-phenotype correlation have only been obtained in mutations found in many patients. The initial studies indicated that mutations in MYH7 would result in severe forms of hypertrophy, and mutations in TNN2 cause a less severe hypertrophy, but a high risk of SCD. Individuals who carry mutations in MYBPC3 would present less severe forms and a lower risk of SCD.3,8,12-25 However, mutations with a poor prognosis have been described in genes initially related to less aggressive forms, a fact that illustrates the difficulty to predict phenotype from genotype. The purpose of our study was to identify the prevalence and phenotypic characteristics of mutations in 5 sarcomeric genes (MYH7,MYBPC3,TNNT2,TNNI3, and TPM1) observed in patients in the Spanish regions of Asturias and Cantabria.
METHODS
Patients
We studied 120 unrelated patients who had been diagnosed between 2002 and 2007 by cardiologists at the Hospital Universitario Central de Asturias and the Hospital Universitario Marqués de Valdecilla in Santander. The diagnosis was performed following the American College of Cardiology/European Society of Cardiology (ACC/ESC) criteria, using as an inclusion criteria a left wall thickness >15 mm on echocardiography when no other cause explained the hypertrophy.26 The clinical characteristics of the patients are shown in Table 1.

Patients with any relative diagnosed with HCM were considered familial cases and patients with no record of any relatives with the condition were considered sporadic cases. In patients with a mutation, the presence of the mutation in all relatives who agreed to participate in the study was determined, regardless of whether they had symptoms of the disease or not; those found to have the mutation underwent an echocardiographic study.
The study was approved by the Clinical Research Ethics Committee of the Hospital Universitario Central de Asturias and all participants gave written informed consent to be included in the study.
Genetic Testing
The polymerase chain reaction was used to amplify the exons and flanking intronic bases of the MYH7(38 exons), MYBPC3 (34 exons), TNNT2 (15 exons), TNNI3 (9 exons), and TPM1 (9 exons) genes. The primers used for the polymerase chain reaction were designed using reference sequences deposited in the GenBank database. Each fragment of the polymerase chain reaction product was purified and sequenced by BigDye chemistry in an ABI310 unit (Applied Biosystems; Foster City, California, United States) (Figure 1). The mutations and polymorphisms found in the 5 sarcomeric genes were named by following the criteria of the Cardiogenomics database (www. cardiogenomics.org). Information on the primers and amplification conditions can be obtained from the authors at the correspondence address.

Figure 1. A: electrophoretic patterns in single-strand conformation analysis gels for the exon 13 fragment of the MYBPC3 gene. Lanes 1, 2, and 6 correspond to normal (unmutated) sequences. In lane 3, the pattern corresponds to the V342D change (mutation); in lane 4, to the R326Q change (polymorphism), and in lane 5, to the A328fs deletion (mutation). B: sequences of fragments with the 3 nucleotide variants of exon 13 of MYBPC3 (*reverse strand sequence).
The mutations in the TNNT2 (15 exons) and TNNI3 (9 exons) genes in 115 of the 120 cases, and in selected exons of MYH7 and MYBPC3 in some patients have already been reported.27-29
Study of Control Subjects
Several conditions must be met to consider a nucleotide change in a gene as a mutation associated with the development of a disease. First, the change must alter the amino acid sequence of the protein. Second, the mutation must be found in all the affected persons of a single family; thus, we can exclude the pathogenic effect of a change if an affected person has not inherited it. Moreover, carriers of the mutation would be more likely to develop the disease and, therefore, should not be found among people with no symptoms of the disease.
All variants found in the patients and not listed in the databases as polymorphisms were analyzed in 200 healthy subjects who had given written informed consent to participate in the study. All were older than 18 years of age and had no symptoms of cardiovascular disease, but had not undergone an echocardiographic study; therefore, asymptomatic hypertrophy cannot be ruled out. Each fragment found to have a nucleotide change that could be a mutation was amplified in the patient who presented it and in the 200 control subjects. The electrophoretic migration pattern was then analyzed in nondenaturing polyacrylamide gels using the single strand conformation analysis (SSCA) technique and following a previously described protocol (Figure 1).27,28
Degree of Amino Acid Conservation Between Species
The mutations affect amino acids that are important for the structure and function of the protein, which would limit its evolutionary divergence. All nucleotide changes that modified the protein sequence and were not found in the control subjects were considered possible pathogenic mutations. As an additional criterion of involvement in the disease, we determined the degree of conservation between humans, chimpanzees, and mice, comparing the sequences of the 3 species deposited in the ENSEMBL database (www.ensembl.org).
Statistical Analysis
The SPSSTM statistical program, version 11.0, was used for the statistical analyses. Analysis of variance (ANOVA) and the Mann-Whitney U test were used to compare continuous variables, and the c2 test was used for the discrete variables. A P value less than .05 was considered significant for all analyses.
RESULTS
In 109 patients, HCM was suspected on the basis of clinical manifestations (exertional dyspnea, palpitations, angina, or syncope) and in 11, because electrocardiographic abnormalities were observed during a routine medical examination. Of the 120 patients, 35 (29%) had at least 1 relative who also had experienced SCD. Thirty-one mutations were found in 32 patients: 10 with mutation in MYH7, 20 in MYBPC3, 2 in TNNT2, and 1 in TPM1 (Tables 2 and 3). Two MYBPC3 mutations (G263X and E542Q) were found in more than 1 patient, 1 had 2 mutations (R278C-TNNT2 and R733H-MYBPC3), and another was homozygous for the A627V mutation in MYBPC3.


Mutations inMYH7
Of the 120 patients, 10 (8%) had gene mutations for beta myosin heavy chain (Table 2). Nine mutations were located in the first 22 exons of the gene, and the K1459N change was found in the protein tail. Four of the mutations have already been described. The mean age at diagnosis of these 10 patients was 35 years (Table 1), and 7 (70%) had a family history of the disease. Patients with the R453C, A583V, and R663H mutation had relatives who had experienced early SCD (before age 50). The V822M mutation was found in a woman diagnosed at 3 years of age. The mutation was not found in either parent and, therefore, would be considered de novo. The patients with R453C and V822M had received transplants at age 43 and 22, respectively.
Mutations in MYBPC3
We found 18 mutations in MYBPC3 in 20 of the 120 (16%) patients (Table 3). Only 6 (33%) of the 18 were known. All MYBPC3 mutations affected amino acid residues that were conserved between species. Five were frameshift changes due to nucleotide insertion/deletion (A328fs del G, Q404fs del C, G532fs del G, M844fs ins GA, and R891fs ins G): 1 was a stop codon and 13 were amino acid changes. The mean age at diagnosis in these patients was 42 years, and 8 (40%) had a family history of HCM and/or SCD (Table 1). In 10 of the sporadic cases, we were able to study some of the relatives and found several asymptomatic carriers (Table 3).
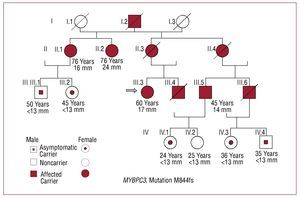
One patient, who was homozygous for the mutation, was the only patient with a mutation in MYBPC3 who had received a heart transplant. Two relatives who carried the mutation were clinically asymptomatic and had no hypertrophy. M844fs was identified in 1 patient with 8 relatives who were also carriers, although only 3 had clinical symptoms of the disease (Figure 2). A patient with the R773H mutation also had the TNNT2-R278C mutation and is described below.

Figure 2. Family tree with M844fs mutation in the MYBPC3 gene. The index case is indicated with an arrow. Three of this patient's relatives were also carriers of the mutation, had clinical symptoms of the disease, and hypertrophy of 14, 17, and 24 mm. Another 5 carriers had no clinical symptoms or hypertrophy.
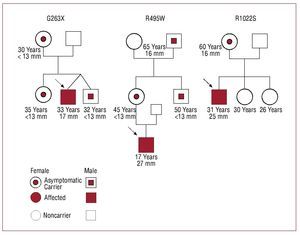
Three young patients experienced exertional dyspnea while engaging in sports (Figure 3). In these cases, mutations G263X, R495W, and R1022S were identified. Patient G263X was 32 years old and had hypertrophy of 19 mm; his mother, sister, and a twin brother also had the mutation, but were clinically asymptomatic and had no hypertrophy. Patient R495W was 17 years old and had a septal wall thickness of 27 mm; his mother, uncle, and grandfather were also carriers, but only the grandfather had hypertrophy (18 mm) at age 85. Patient R1022S was 31 years old and had a septum of 25 mm; his mother also had the mutation, but showed no clinical symptoms, except for a septum of 18 mm.

Figure 3. Families of 3 sporadic cases with mutations in MYBPC3. The index cases are indicated with an arrow. All 3 were regularly involved in sports and were diagnosed after presenting symptoms associated with physical exercise. In the families of the R495WS and R1022S patients, there was another carrier without symptoms of the disease, but with cardiac hypertrophy.
Mutations in TNNT2, TNNI3, and TPM1
In 2 (1.66%) of the 120 patients, we found the R92Q and R278C mutations in TNNT2 (Table 2). The case with R278C also had the R733H mutation in MYBPC3. In the TPM1 gene, the D175N mutation was found in 1 patient diagnosed at age 41 with severe hypertrophy (32 mm) (Table 2). His son and his brother had this mutation and a hypertrophy of 27 and 20 mm.
Double Mutants
A woman diagnosed at age 49 had 2 mutations: R278C in TNNT2 and R733H in MYBPC3. A 40-year-old daughter and a 6-year-old granddaughter were double carriers, but were asymptomatic; 2 daughters age 47 and 42 were carriers of the R733H mutation, but also had no clinical symptoms or hypertrophy. A 52-year-old sister with the R278C mutation had mild symptoms and a septal thickness of 13 mm.
Genotype-Phenotype Correlation
The clinical and echocardiographic characteristics were compared according to mutated gene (Table 1). The mean age at diagnosis was lower in patients with mutations in MYH7 compared with MYBPC3, although the difference was not significant. Patients with no mutations were older on average than patients with mutations, although the differences were not significant. Hypertrophy was 21 (5) mm in the MYH7 patients, 22 (5) mm in the MYBPC3patients, and 19 (6) mm in patients without a mutation. A history of the disease was reported by 70% of MYH7 patients, compared with 40% of MYBPC3 patients and 18% of patients with no mutation.
Polymorphisms
In addition to the mutations, various changes unrelated to the disease (polymorphisms) were found in the 5 genes. All these nucleotide changes were also identified in the control subjects. In MYH7, 27 polymorphisms were found; 23 were in the exons and only 1 involved an amino acid change (S1491C). In MYBPC3, 24 polymorphisms were found, 11 of them in exons, of which 5 showed amino acid changes: R17Q, S236G, R326Q, W382R, and V896M. Information on the changes in these genes can be requested from the authors at the correspondence address.
DISCUSSION
Our study is the first to analyze the complete sequence of the 5 most commonly mutated sarcomeric genes in HCM in a large series of Spanish patients. Previous reports have investigated all the MYH7exons in large series and several exons of various genes in small case studies.27-32
The most commonly mutated gene was MYBPC3(16% of cases), followed by MYH7 (8%), and TNNT2and TPM1 (<2%). We found a lower incidence of mutations, compared to those described by other authors. Most of those studies were conducted at referral hospitals that had received patients with severe forms of HCM who would have been more likely to have a family history of the disease. In our study, only 29% of cases had a history of HCM and/ or SCD, whereas other studies had up to 90% of familial forms.8,12 The frequency of sarcomeric mutations would be higher among patients with a family history of the disease, and the lower number of mutations identified in our study could be due to a higher frequency of sporadic cases. Additionally, no mutations were found in 43% of patients with a family history of HCM, which indicates that other genes could explain the familial segregation in these cases.
The low frequency of mutations in MYH7 (8%) has been described by other authors, such as Laredo et al30 in patients from Galicia. Of the patients with MYBPC3 mutations, 61% were sporadic, compared with only 30% of those with a MYH7 mutation. Higher penetration has been reported3,8,12,15,33 for MYH7 mutations, which would increase the probability that the disease will manifest in the carriers of each family. Patients with MYH7mutations would manifest the disease at an earlier age and would have a higher degree of hypertrophy, a more malignant phenotype, and a poorer prognosis.8 Although we found a younger age at onset of the disease among MYH7 patients, the difference with MYBPC3 patients and cases with no mutation was not statistically significant, probably because of the smaller number of patients with mutations. Moreover, no differences were found in the interventricular septal thickness between the 3 groups. The low frequency of mutations in the thin-filament encoding genes TNNT2 and TPM1(<2%) is similar to that described by other groups.8
The initial classification of the mutations as "malignant" or "benign" has been refined in more recent studies that have shown the difficulty of stratifying the prognosis for most mutations.8,12-14 Two of our cases illustrate this clinical heterogeneity, even among carriers in the same family. A woman with 2 mutations in TNN2 and MYBPC3 had been diagnosed due to angina and dyspnea at age 49 and had hypertrophy of 22 mm. A daughter and a granddaughter were also double carriers, but were clinically asymptomatic and had no hypertrophy. This indicates that the presence of 2 sarcomeric mutations would not necessarily be associated with an early, severe manifestation of the disease. In another family with the M844fs mutation in MYBPC3, 9 carriers were identified, of which only 3 had hypertrophy. Some carriers remained asymptomatic into an advanced age, but 2 had suffered sudden cardiac death before age 50. These cases indicate that the clinical manifestations are a result of genetic and nongenetic risk factors and, therefore, the genetic information of each patient should not be used as the only basis to establish the prognosis. Nevertheless, individuals with no heart disease but carriers of sarcomeric gene mutations could manifest symptoms at a later date and, therefore, should undergo periodic assessments to avoid the adverse effects of HCM.
Three patients were diagnosed by ultrasound, performed to investigate fatigue associated with physical exercise. The genetic study identified 3 mutations in MYBPC3 in their 3 families, in which we found asymptomatic carriers. These 3 cases indicate that some mutations, particularly in MYBPC3, could have low penetration, but that physical exercise would accelerate the development of symptoms and hypertrophy among the carriers. In athletes with no history of the disease, HCM may indicate the presence of a sarcomeric mutation, probably in MYBPC3.
All mutations in MYH7 would translate into changes in a single amino acid, whereas MYBPC3also had frameshift changes. Moreover, polymorphisms with an amino acid change were less common in MYH7. This indicates selective pressure against mutations that modify various amino acids of the beta myosin heavy chain that would lead to a high risk of early death; hence, they would not be observed in adults with HCM.8
Finally, half the mutations in MYH7 (5/10) and most of those found in MYBPC3 (13/18) have not been previously described.33 Only 2 of the 11 mutations found by Laredo et al30 (R663H and K1459N) were also found in our patients. This indicates that direct analyses of known mutations would not be highly useful because they do not identify mutations in true carrier cases.34 Complete sequencing of the sarcomeric genes is necessary in these cases to definitively exclude the presence of any mutation.
CONCLUSIONS
In an analysis of the 5 most commonly mutated sarcomeric genes in HCM in a series of 120 patients in the regions of Asturias and Cantabria, mutations were found in 26% of cases. The most commonly mutated gene was MYBPC3, followed by MYH7, TNNT2, and TPM1. More than half the mutations have not been described. We found no mutations in TNNI3. There were no differences in the mean age at diagnosis or in the interventricular septal thickness between the MYH7 and MYBPC3 carriers. Our study illustrates the difficulty to define the prognosis in carriers with mutations in these genes.
ACKNOWLEDGEMENTS
We wish to express our appreciation to the patients and their relatives.
ABBREVIATIONS
ANOVA: analysis of variance
HCM: hypertrophic cardiomyopathy
SCD: sudden cardiac death
SSCA: single strand conformation analysis
Study funded by the FIS 06/0214 project of the Fondo de Investigaciones Sanitarias (Health Research Fund)-ERDF European Regional Development Fund (principal investigator, Eliecer Coto García). Red de Investigación Renal (Renal Research Network) REDINREN [RD06/0016]) of the Instituto de Salud Carlos III. Eliecer Coto is a fellow of the Research Activity Enhancement Program of the Instituto de Salud Carlos III.
Correspondence: Dr E. Coto.
Laboratorio de Genética Molecular. Hospital Universitario Central de Asturias (Maternidad).
33006 Oviedo. Asturias. España.
E-mail: eliecer.coto@sespa.princast.es
Received April 28, 2008.
Accepted for publication September 12, 2008.