Catecholaminergic polymorphic ventricular tachycardia is a malignant disease, due to mutations in proteins controlling Ca2+ homeostasis. While the phenotype is characterized by polymorphic ventricular arrhythmias under stress, supraventricular arrhythmias may occur and are not fully characterized.
MethodsTwenty-five relatives from a Spanish family with several sudden deaths were evaluated with electrocardiogram, exercise testing, and optional epinephrine challenge. Selective RyR2 sequencing in an affected individual and cascade screening in the rest of the family was offered. The RyR2R420Q mutation was generated in HEK-293 cells using site-directed mutagenesis to conduct in vitro functional studies.
ResultsThe exercise testing unmasked catecholaminergic polymorphic ventricular tachycardia in 8 relatives (sensitivity = 89%; positive predictive value = 100%; negative predictive value = 93%), all of them carrying the heterozygous RyR2R420Q mutation, which was also present in the proband and a young girl without exercise testing, a 91% penetrance at the end of the follow-up. Remarkably, sinus bradycardia, atrial and junctional arrhythmias, and/or giant post-effort U-waves were identified in patients. Upon permeabilization and in intact cells, the RyR2R420Q expressing cells showed a smaller peak of Ca2+ release than RyR2 wild-type cells. However, at physiologic intracellular Ca2+ concentration, equivalent to the diastolic cytosolic concentration, the RyR2R420Q released more Ca2+ and oscillated faster than RyR2 wild-type cells.
ConclusionsThe missense RyR2R420Q mutation was identified in the N-terminus of the RyR2 gene in this highly symptomatic family. Remarkably, this mutation is associated with sinus bradycardia, atrial and junctional arrhythmias, and giant U-waves. Collectively, functional heterologous expression studies suggest that the RyR2R420Q behaves as an aberrant channel, as a loss- or gain-of-function mutation depending on cytosolic intracellular Ca2+ concentration.
Keywords
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is responsible for 12% to 56% of sudden cardiac deaths or cardiac arrests with structurally normal heart.1,2 Catecholaminergic polymorphic ventricular tachycardia manifests as syncope or sudden cardiac death triggered by adrenergic states with an estimated 80% penetrance.3 Resting electrocardiogram is usually normal and patients develop ventricular arrhythmias (VA) during exercise testing (ET) or catecholamine infusion.1,4 The pathogenesis and real prevalence of non-VA, which have occasionally been reported in CPVT patients remains more elusive, including sinus dysfunction, wandering atrial pacemaker, junctional arrhythmias, and atrial fibrillation and flutter.3,5–9
Mutations usually affect the gene encoding the cardiac ryanodine receptor (RyR2), but other genes have also been involved.10–14 Abnormal intracellular calcium (Ca2+) handling underlies an increased diastolic Ca2+ release, delayed after depolarizations and triggered activity, which is the pathophysiological basis of VA in this disease.3,15 However, the precise mechanisms might differ depending on the specific mutation at the RyR2 protein.16–18 Herein we present an exhaustive characterization of a large CPVT family, stressing the electrocardiographic features of the disease. Moreover, in vitro insights from the mechanism of RyR2R420Q channel dysfunction are provided.
METHODSA family with 4 cases of sudden death underwent familial evaluation with a protocol conforming to the Declaration of Helsinki and previously approved by the local research ethics committee. Informed consent was obtained from each individual.
Clinical Work-upElectrocardiogram, echocardiography blood sampling, and maximal ET (Bruce protocol) were performed in individual III:10. Once the CPVT phenotype was unmasked during the ET, cascade screening was offered, including an epinephrine challenge to adults with unremarkable ET who were offspring of an affected individual. Maximal U-wave to T-wave amplitude ratio on exertion was recorded. Sinus bradycardia was defined as a heart rate<60 bpm over 14 years of age or lower than percentile 2 adjusted to age in younger children.7 Catecholaminergic polymorphic ventricular tachycardia was diagnosed provided sudden cardiac death, polymorphic VA or frequent premature ventricular contractions (> 10/min) during ET, or epinephrine challenge were present (phenotype-positive).19
Genetic Work-upDNA was obtained from whole blood (relatives) or from paraffin-embedded myocardium (proband). Targeted mutational RyR2 analysis (direct sequencing of exons 3, 8, 14, 15, 37, 44-50, 83, 87-105, and adjacent intronic regions, GenBank accession number NM_001035) with an ABI Prism 3100 sequencer (Applied Biosystems) in individual III:10 identified the RyR2R420Q mutation and exon 14 was sequenced in the remaining relatives (mutation carriers were considered genotype+). Since Andersen Tawil syndrome is characterized by a normal or near-normal QTc, giant U waves and polymorphic exercise-related VA due to mutations in KCNJ2 gene, this gene was also sequenced in CPVT individuals.
Generation of RyR2 Wild Type and RyR2R420Q ConstructsRyR2 site-directed mutagenesis was performed to create RyR2R420Q.20 Briefly, a plasmid encoding human cardiac N-terminal RyR (Genbank X98330) was used for site-directed mutagenesis (Stratagene, ChickChange kit) using the oligonucleotide 5’-CAGCCCGAGTTATCCAGAGCACAGTCTTCC-3’. The final construct pRyRR420Q was generated by SanDI/SpeI digestion and inserted into a pcDNA3 (Invitrogen) containing full-length, RyR2-enhanced, green fluorescent protein.
Western Blot AnalysisHEK-293 cells (European Collection of Cell Culture Agency, Salisbury, United Kingdom) were transfected.20–22 Protein fractions (100μg) were resuspended in SDS-PAGE loading buffer, and proteins were separated in a 4% SDS-PAGE gel strengthened with 0.5% agarose (for RyR).21 Proteins from 4% gels were electrophoretically transferred to a polyvinylidenedifluoride membrane (Immobilon-P, Millipore) using a semi-dry transfer system (Trans-Blot SD, Bio-Rad) at 22V for 4h. Primary antibody specific for the RyR2 isoform (Ab1093, rabbit polyclonal antibody raised to RyR2 residues 4454-4474) was applied at 1:1000 dilution overnight at 4°C. Immunoreactive protein bands were visualized by enhanced chemiluminescence detection (ECL, Amersham Biosciences).
Intracellular Ca2+ ImagingHEK-293 cells were plated in polylysineD-coated, glass-bottom Petri dishes (MatTek; Ashland, United States) and transfected with enhanced green fluorescent protein-tagged RyR2R420Q and RyR2WT plasmids.20,22 Cells were permeabilized to control [Ca2+]i with saponin (0.01%).12 The internal solution contained (mmol/L): 120 K-aspartate, 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, 3 Mg-adenosine triphosphate, 0.5 EGTA (ethylene glycol tetraacetic acid), 10 Na phosphocreatine, 5 U/mL creatine phosphokinase, 0.75 MgCl2 and 8% dextran, saponin (0.01%), pH 7.2. After permeabilization, cells were perfused with the same internal solution without saponin but with 15μM fluo-4 pentapotassium salt and known [Ca2+] (10-1–105nM) obtained using different CaCl2:EGTA ratios calculated with Maxchelator.23 The EGTA concentration was constant at 0.5mM. Because HEK-293 cells are not excitable, RyR2 was activated by 5mM caffeine and the resulting [Ca2+]i transients were recorded using confocal microscopy (Zeiss LSM 510 water-immersion objective ×63; numerical aperture 1.2) at 394ms/frame. We also performed experiments in intact HEK-293 cells as described for other RyR2 constructs.24,25 In this case, the cells loaded with fluo-4 AM (acetoxymethyl ester). Afterwards, cells were perfused with external solution containing (Mm): 150 NaCl, 5.4 KCl, 1.5 CaCl2, 1 MgCl2, 10 glucose, 5 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (pH 7.4). Spontaneous [Ca2+]i oscillations were recorded using confocal microscopy. In both permeabilized and intact cells, fluo-4 fluorescence was excited by an Ar laser (488nm) and emission fluorescence collected at>505nm. Image analyses were performed by Zeiss LSM 510 v2.8 software. Enhanced green fluorescent protein background fluorescence was subtracted. The F (fluorescence) values were normalized by the F0 (basal fluorescence, determined before the application of the caffeine or in between 2 oscillations) in order to obtain the F/F0 (fluorescence ratio).
Statistical AnalysisClinical continuous variables were expressed as mean (standard deviation), laboratory, continuous variables as mean (standard error mean), and Student t tests for comparison between groups were applied. Dichotomous variables were expressed as percentages and the chi-square test for comparison between groups was applied (Fisher exact correction, when applicable). A 2-tailed P value < .05 was considered statistically significant. In the analyses, SPSS 12.0 software (SPSS, Inc.; Chicago, Illinois, United States) was used.
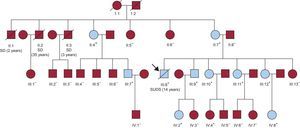
RESULTSFamily History of Sudden Cardiac DeathThe proband (Figure 1, III:8), a 14-year-old male with previous history of unexplained exertion syncopes, suffered a sports-related sudden death with an unremarkable postmortem. Three more relatives had also died suddenly and young (one of them, II:2, during exertion and with a previous unexplained syncope), but no autopsy was performed in those cases.
 at the end of the follow-up. The arrow points to the proband. Plus and minus signs depict mutation-positive and mutation-negative individuals, respectively. SCD: sudden cardiac death with postmortem without structural heart disease; SD: sudden death, no autopsy.")
Family pedigree. Squares, males. Circles, females. Crossed symbols, deceased individuals. Blue symbols, catecholaminergic polymorphic ventricular tachycardia phenotype+ (with sudden cardiac death or ventricular effort-related arrhythmias) at the end of the follow-up. The arrow points to the proband. Plus and minus signs depict mutation-positive and mutation-negative individuals, respectively. SCD: sudden cardiac death with postmortem without structural heart disease; SD: sudden death, no autopsy.
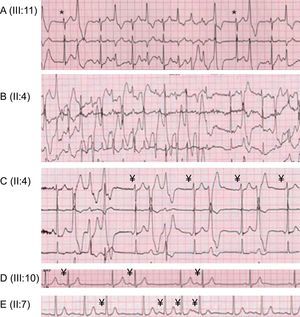
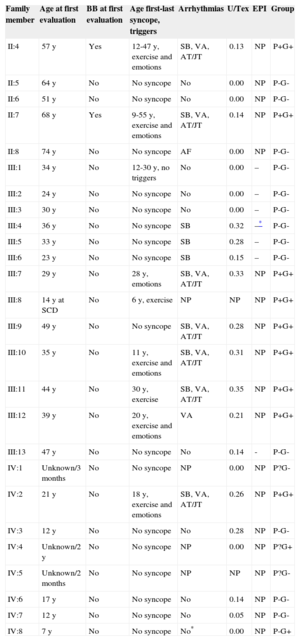
Fifteen years following the index event, 25 living proband relatives underwent evaluation (Table). Structural heart disease was ruled out with routine echocardiography and, for patient III:10, also with cardiac magnetic resonance imaging. Three relatives could not be evaluated with a routine ET because of their young age (unknown phenotype). Among them, only one (IV:4) was proven to be genotype+ and electrocardiogram monitoring did not show VA (either without drugs at the initial evaluation at 2 years old or on weight-adjusted beta-blockers at 4 years old). In the remaining 22 relatives, ET allowed the clinical diagnosis of CPVT in 8 patients (phenotype+). Typically, a variable increase in heart rate was followed by a progressive appearance of premature ventricular contractions at a mean heart rate threshold of 103 (24) [62-130] bpm, first isolated and monomorphic and then in bigeminy, polymorphic, in couplets, and also in bursts of nonsustained polymorphic ventricular tachycardia in 50% of phenotype+ individuals at a mean heart rate threshold of 121 (10) [109-131] bpm. Finally, premature ventricular contractions progressively disappeared during the recovery period. Only one individual showed the bidirectional ventricular tachycardia. Of note, these arrhythmias were neither sustained nor syncopal (Figure 2). Among the 13 individuals at risk, the epinephrine challenge was negative in 7 individuals, in keeping with the previous negative ETs, and was declined in 4 children (IV:3 and IV:6-8) and 2 adults (II:5 and II:6), awaiting the genetic results.
Initial Familial Work-up
| Family member | Age at first evaluation | BB at first evaluation | Age first-last syncope, triggers | Arrhythmias | U/Tex | EPI | Group |
|---|---|---|---|---|---|---|---|
| II:4 | 57 y | Yes | 12-47 y, exercise and emotions | SB, VA, AT/JT | 0.13 | NP | P+G+ |
| II:5 | 64 y | No | No syncope | No | 0.00 | NP | P-G- |
| II:6 | 51 y | No | No syncope | No | 0.00 | NP | P-G- |
| II:7 | 68 y | Yes | 9-55 y, exercise and emotions | SB, VA, AT/JT | 0.14 | NP | P+G+ |
| II:8 | 74 y | No | No syncope | AF | 0.00 | NP | P-G- |
| III:1 | 34 y | No | 12-30 y, no triggers | No | 0.00 | – | P-G- |
| III:2 | 24 y | No | No syncope | No | 0.00 | – | P-G- |
| III:3 | 30 y | No | No syncope | No | 0.00 | – | P-G- |
| III:4 | 36 y | No | No syncope | SB | 0.32 | –* | P-G- |
| III:5 | 33 y | No | No syncope | SB | 0.28 | – | P-G- |
| III:6 | 23 y | No | No syncope | SB | 0.15 | – | P-G- |
| III:7 | 29 y | No | 28 y, emotions | SB, VA, AT/JT | 0.33 | NP | P+G+ |
| III:8 | 14 y at SCD | No | 6 y, exercise | NP | NP | NP | P+G+ |
| III:9 | 49 y | No | No syncope | SB, VA, AT/JT | 0.28 | NP | P+G+ |
| III:10 | 35 y | No | 11 y, exercise and emotions | SB, VA, AT/JT | 0.31 | NP | P+G+ |
| III:11 | 44 y | No | 30 y, exercise | SB, VA, AT/JT | 0.35 | NP | P+G+ |
| III:12 | 39 y | No | 20 y, exercise and emotions | VA | 0.21 | NP | P+G+ |
| III:13 | 47 y | No | No syncope | No | 0.14 | - | P-G- |
| IV:1 | Unknown/3 months | No | No syncope | NP | 0.00 | NP | P?G- |
| IV:2 | 21 y | No | 18 y, exercise and emotions | SB, VA, AT/JT | 0.26 | NP | P+G+ |
| IV:3 | 12 y | No | No syncope | No | 0.28 | NP | P-G- |
| IV:4 | Unknown/2 y | No | No syncope | NP | 0.00 | NP | P?G+ |
| IV:5 | Unknown/2 months | No | No syncope | NP | NP | NP | P?G- |
| IV:6 | 17 y | No | No syncope | No | 0.14 | NP | P-G- |
| IV:7 | 12 y | No | No syncope | No | 0.05 | NP | P-G- |
| IV:8 | 7 y | No | No syncope | No* | 0.00 | NP | P-G+ |
+, positive; –, negative; ?, unknown; AT/JT, atrial and/or junctional tachyarrhythmias; BB, beta-blockers; EPI, epinephrine challenge; ET, exercise testing; P, catecholaminergic polymorphic ventricular tachycardia phenotype; G, RyR2R420Q genotype; NP, not performed; SB, sinus bradycardia; SCD, sudden cardiac death; U/Tex, maximal U-wave to T-wave amplitude ratio on exertion; VA, ventricular arrhythmias.
. B: bidirectional nonsustained ventricular tachycardia. C: ventricular premature beats, nonsustained polymorphic ventricular tachycardia, polymorphic ventricular couplet, monomorphic ventricular bigeminy, and atrial ectopic rhythm (¥). D: atrial bigeminy (¥). E: atrial premature beats isolated and in salvos (¥).")
Ventricular and nonventricular arrhythmias during exercise testing. A: ventricular monomorphic bigeminy and bursts of nonsustained polymorphic ventricular tachycardia (*junctional escape). B: bidirectional nonsustained ventricular tachycardia. C: ventricular premature beats, nonsustained polymorphic ventricular tachycardia, polymorphic ventricular couplet, monomorphic ventricular bigeminy, and atrial ectopic rhythm (¥). D: atrial bigeminy (¥). E: atrial premature beats isolated and in salvos (¥).
A heterozygous missense mutation in RyR2 (1259 G>A, R420Q) was identified in individual III:10. Cascade screening for this mutation rendered a total of 11 genotype+ individuals: the 8 phenotype+ individuals, 1 young phenotype – girl (IV:8), 1 young unknown phenotype girl (IV:4), and also the proband (III:8). All family members were classified according to their phenotype and genotype (+, –, or unknown) (Table). No KCNJ2 mutations were found.
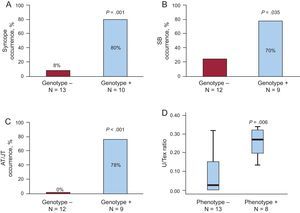
Considering patient status as a mutation carrier, ET at first evaluation showed a sensitivity of 89%, a positive predictive value of 100%, and a negative predictive value of 93%. The only false negative ET was obtained in a young asymptomatic genotype+ girl (IV:8) aged 7 years who converted into phenotype+ despite beta-blockers 2 years thereafter. Consequently, overall performance of the ET increased up to 100%. Disease penetrance increased from 82% to 91% at the end of follow-up, when clinical (syncope and/or sudden cardiac death) and ET results were considered in genotype+ patients. Notably, syncope occurrence was associated with mutation carrier status in assessable individuals>6 years (8/10 genotype+ with stress-triggers vs 1/13 genotype–individuals without stress-triggers; P = .001) (Table, Figure 3A).

Clinical features. A: syncope occurrence. Children under the age of the first syncope in the family, 6 years old, were not considered. B: sinus bradycardia occurrence. C: atrial and junctional arrhythmias. D: maximal U-wave to T-wave amplitude ratio on exertion comparison attending to the phenotype. For B-D, only individuals in sinus rhythm and with available exercise testing were considered. +, positive; –, negative; AT/JT, atrial and/or junctional tachyarrhythmias; SB, sinus bradycardia; U/Tex, maximal U-wave to T-wave amplitude ratio on exertion.
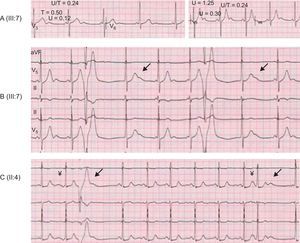
Sinus, atrial, junctional, and VA were identified in CPVT patients both in resting and exercise electrocardiograms. Assessable individuals (> 6 years old, in sinus rhythm, and with available ET) were evaluated with respect to non-VA (N = 21; 9 genotype+, 12 genotype-). A higher incidence of sinus bradycardia (Table, Figure 3B) was observed in genotype+ patients when compared with genotype– individuals (78% vs 25%, odds ratio=10.5; P = .030). Atrial and/or junctional tachyarrhythmias included atrial premature beats, atrial bigeminy, nonsustained atrial tachycardia, atrial and junctional accelerated rhythm, and junctional escapes. Atrial and/or junctional tachyarrhythmias were detected in 7/9 genotype+ patients and in none of the genotype– individuals (Table, Figure 3C). Maximal U-wave to T-wave amplitude ratio on exertion was significantly higher in phenotype+ than in phenotype– individuals (Figure 3D), although data overlap precluded the identification of an accurate cut-off point. Remarkably, giant postexercise U-waves were seen in several CPVT patients (Figure 4). Neither U-wave alternans nor post-extrasystolic U-wave polarity changes were detected.
 and on exertion (right). A fixed, short PR interval suggests an accelerated atrioventricular conduction (left). B: transient increase in U-wave amplitude in the first beat after a ventricular premature beat (arrows) on exertion. A junctional rhythm competes with sinus rhythm. C: transient increase in U-wave amplitude on exertion associated to premature beats, either atrial (right arrow) or ventricular (left arrow).")
U-wave features in catecholaminergic polymorphic ventricular tachycardia patients. A: resting U-wave (left) and on exertion (right). A fixed, short PR interval suggests an accelerated atrioventricular conduction (left). B: transient increase in U-wave amplitude in the first beat after a ventricular premature beat (arrows) on exertion. A junctional rhythm competes with sinus rhythm. C: transient increase in U-wave amplitude on exertion associated to premature beats, either atrial (right arrow) or ventricular (left arrow).
Maximal tolerated dose of beta-blockers was achieved in genotype+ individuals. Five implantable cardioverter defibrillators were placed in phenotype+ patients with frequent VA despite maximal treatment with beta-blockers (one of them, IV:2, also reported stress-triggered presyncopes). At that time, reports concerning the role of flecainide in CPVT had not yet been published4. No implantable cardioverter defibrillator-shock has been registered yet (mean follow-up 23.3 months).
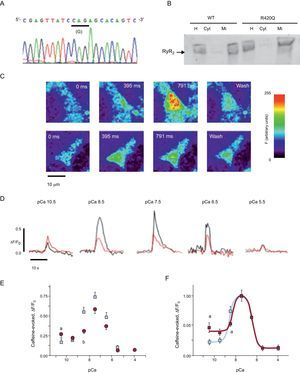
In Vitro RyR2R420Q Cell ModelTo test whether RyR2R420Q channel function was altered, we generated the mutation and expressed it in HEK-293 cells (Figure 5A). The expressed RyR2-specific immunoreactive proteins were located at the sarcoplasmic reticulum membrane (Figure 5B). Figure 5C depicts examples of confocal images in permeabilized HEK-293 cells expressing RyR2WT and RyR2R420Q under caffeine addition at [Ca2+]i10–7.5 M. Additionally, pictures of fluorescence profiles recorded at various [Ca2+]i are plotted in Figure 5D. Both RyR2WT and RyR2R420Q Ca2+ releases displayed a bell-shaped curve when plotted as a function of cytosolic [Ca2+]i with a 34% reduction in the peak of Ca2+ release in RyR2R420Q expressing cells (Figure 5E) at pCa 7.5. A different degree of RyR2 expression in each cell group was ruled out as an explanation for this finding when a similar enhanced green fluorescent protein-RyR2 fluorescence was measured in RyR2WT and RyR2R420Q cells (F, in arbitrary units, 50.4 (1.6) in 195 RyR2WTcells vs 50.1 (1.5) in 241 RyR2R420Q expressing cells; no significant difference). However, at the lowest pCa tested, a significant increase in caffeine-evoked Ca2+ release was observed in RyR2R420Q cells (pCa10.5, Figures 5D and 5E). In order to calculate the EC50 for the Ca2+ activating curve and the IC50 (half maximal inhibitory concentration) for the Ca2+ inactivating curve, we normalized the caffeine-evoked fluorescence by its peak, which happened in both groups at the same [Ca2+]i (pCa=7.5) (Figure 5F). The EC50 (half maximal effective concentration) was lower in RyR2R420Q when compared to RyR2WT (2.66 [0.13] in RyR2R420Q vs 4.98 [5.15]nM in RyR2WT; no significant difference). At higher [Ca2+]i the RyR2 was inactivated with a similar pattern, as observed in the descending portion of the fitted curves (IC50, 365.5 (50.5) in RyR2WT vs 339.5 (58.4) nM in RyR2R420Q; no significant difference).
![Functional in vitro assessment of the RyR2R420Qmutation. A: electropherogram confirming the introduction of the point mutation G1380A that results in conversion of arginine (A) to glutamine (G), RyR2R420Q, in the plasmid contruct. B: Western blot analysis of RyR2 from transfected HEK-293 cells confirming that the RyR2RWT and RyR2R420Q proteins are expressed in the homogenate and the microsomal fractions, but not in the cytosol. The arrow points the 595kDa band (RyR2-enhanced green fluorescent protein). C: confocal images of calcium changes in permeabilized HEK-293 cells expressing RyR2WT (top) and RyR2R420Q (bottom) and superfused with internal solution containing [Ca2+]I = 10–7.5M and 5mM caffeine. D: representative examples of fluorescence profiles obtained from permeabilized HEK-293 cells expressing RyR2WT (black) and RyR2R420Q (red) and superfused with solutions containing different [Ca2+]i and stimulated with 5mM caffeine. E: averaged caffeine-induced calcium release (peak values normalized for the basal fluorescence ratio) in HEK-293 cells expressing RyR2WT (squares) and RyR2R420Q (circles) at various [Ca2+]i. Data are expressed as mean (standard error of the mean), N = 20-50 cells per group. F: averaged enhanced green fluorescent protein-RyR2 fluorescence observed in HEK-293 cells expressing RyR2WT (N = 195) and RyR2R420Q (N = 241). △, increase; Cyt, cytosol; eGFP, enhanced green fluorescent protein; F, fluorescence; F0, basal fluorescence; H, homogenate; Mi, microsomal fractions. aP<.05, RyR2R420Q vs RyR2WT. bP<.001.](https://static.elsevier.es/multimedia/18855857/0000006800000005/v1_201504231305/S1885585714003314/v1_201504231305/en/main.assets/gr5.jpeg?xkr=eyJpdiI6IjVQK0FFRGlOL0tBeXZMUTZzVzlBMVE9PSIsInZhbHVlIjoiVVFuWjRXSkwxMzYxd2EyMVF4L3hhcVI4cjIvMEFaTkdYZFI2S2NscGQrNlp4Z2FZdlQ0QytVOGJzR1QyNWdLM1phUno1OFV0bmh1RDZ5OGpsSlhlckUwTzRkcGo1eDZFK2F2Yjkza1JVdldteDAyU3UrS0xBYkdyRXZ5b1RXakFOaFh0Nll3S3IycitDRnNsUExGSHJYU1pOWmpxQ2huSGJBaXRiTHFCZnhtYXdWRDNBczllcW5DNVNzUEI3cHZuamxHa3hxVDd1UnZzeDZTeElTRzVtMUtmcnA5WkxPR1ZHTXlMRzlkVTF4ZHBOdVY3M3MyYmx6a2tEQjF5V01Yd3I2K1FqTHlSdU1IR2VpbW9JZUNKd09CZVczaDBFWWpSTFRGSXBYU095ZDA9IiwibWFjIjoiZmU4OGFkNDAxNWJhODI4YmM4NDQwNjlkYjk2YTk4ZjI2NGNkYzcyOTNkNmJlYjQ5NTM3ZTJjNWMyYjE4OTM2YyIsInRhZyI6IiJ9 "Functional in vitro assessment of the RyR2R420Qmutation. A: electropherogram confirming the introduction of the point mutation G1380A that results in conversion of arginine (A) to glutamine (G), RyR2R420Q, in the plasmid contruct. B: Western blot analysis of RyR2 from transfected HEK-293 cells confirming that the RyR2RWT and RyR2R420Q proteins are expressed in the homogenate and the microsomal fractions, but not in the cytosol. The arrow points the 595kDa band (RyR2-enhanced green fluorescent protein). C: confocal images of calcium changes in permeabilized HEK-293 cells expressing RyR2WT (top) and RyR2R420Q (bottom) and superfused with internal solution containing [Ca2+]I = 10–7.5M and 5mM caffeine. D: representative examples of fluorescence profiles obtained from permeabilized HEK-293 cells expressing RyR2WT (black) and RyR2R420Q (red) and superfused with solutions containing different [Ca2+]i and stimulated with 5mM caffeine. E: averaged caffeine-induced calcium release (peak values normalized for the basal fluorescence ratio) in HEK-293 cells expressing RyR2WT (squares) and RyR2R420Q (circles) at various [Ca2+]i. Data are expressed as mean (standard error of the mean), N = 20-50 cells per group. F: averaged enhanced green fluorescent protein-RyR2 fluorescence observed in HEK-293 cells expressing RyR2WT (N = 195) and RyR2R420Q (N = 241). △, increase; Cyt, cytosol; eGFP, enhanced green fluorescent protein; F, fluorescence; F0, basal fluorescence; H, homogenate; Mi, microsomal fractions. aP<.05, RyR2R420Q vs RyR2WT. bP<.001.")
Functional in vitro assessment of the RyR2R420Qmutation. A: electropherogram confirming the introduction of the point mutation G1380A that results in conversion of arginine (A) to glutamine (G), RyR2R420Q, in the plasmid contruct. B: Western blot analysis of RyR2 from transfected HEK-293 cells confirming that the RyR2RWT and RyR2R420Q proteins are expressed in the homogenate and the microsomal fractions, but not in the cytosol. The arrow points the 595kDa band (RyR2-enhanced green fluorescent protein). C: confocal images of calcium changes in permeabilized HEK-293 cells expressing RyR2WT (top) and RyR2R420Q (bottom) and superfused with internal solution containing [Ca2+]I = 10–7.5M and 5mM caffeine. D: representative examples of fluorescence profiles obtained from permeabilized HEK-293 cells expressing RyR2WT (black) and RyR2R420Q (red) and superfused with solutions containing different [Ca2+]i and stimulated with 5mM caffeine. E: averaged caffeine-induced calcium release (peak values normalized for the basal fluorescence ratio) in HEK-293 cells expressing RyR2WT (squares) and RyR2R420Q (circles) at various [Ca2+]i. Data are expressed as mean (standard error of the mean), N = 20-50 cells per group. F: averaged enhanced green fluorescent protein-RyR2 fluorescence observed in HEK-293 cells expressing RyR2WT (N = 195) and RyR2R420Q (N = 241). △, increase; Cyt, cytosol; eGFP, enhanced green fluorescent protein; F, fluorescence; F0, basal fluorescence; H, homogenate; Mi, microsomal fractions. aP<.05, RyR2R420Q vs RyR2WT. bP<.001.
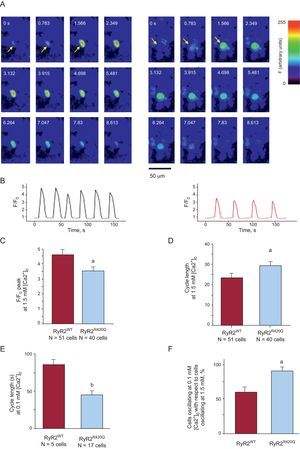
Intact HEK-293 cells transfected with RyR2WT or RyR2R420Q plasmids were used to analyze spontaneous [Ca2+]i oscillations (Figures 6A and 6B). Interestingly, at physiological [Ca2+]o (1.5mM) [Ca2+]i oscillations recorded in RyR2R420Q cells had lower amplitude (Figure 6C), shorter duration (7.6 [0.5] ms in 51 RyR2WT cells, vs 5.9 [0.5] ms in 40 RyR2R420Q cells; P < .05), and longer cycle length than in RyR2WT cells (Figure 6D). Notably, these alterations were not due to a decrease in the sarcoplasmic reticulum Ca2+ load, which was in fact similar between both cell groups (F/F0, 3.8 [0.3] in 15 RyR2WTcells vs 3.6 [0.3] in 13 RyR2R420Q cells, no significant difference). In a group of cells, we first recorded spontaneous oscillations at 1.5mM [Ca2+]o and then lowered [Ca2+]o to 0.1mM [Ca2+]o. This maneuver suppressed the automatic activity in a greater proportion of RyR2WT than of RyR2R420Q HEK-293 cells, as evidenced by a higher frequency (or shorter cycle length) of spontaneous oscillations (Figure 6E) and a higher proportion of oscillating RyR2R420Q expressing cells (Figure 6F), in keeping with our findings in permeabilized cells at the lowest [Ca2+]i analyzed.
![Spontaneous Ca2+ oscillations in HEK-293 cells expressing RyR2R420Q are smaller and slower but change to faster than RyR2WT at lower [Ca2+]o. A: time series of confocal images in intact HEK-293 cells expressing RyR2WT (left) and RyR2R420Q (right). B: representative fluorescence profiles in intact HEK-293 cells expressing RyR2WT (left) and RyR2R420Q (right), at 1.5mM [Ca2+]o. C: amplitude of Ca2+ oscillations at 1.5mM [Ca2+]o. D: as in C but for the cycle length between consecutive Ca2+ oscillations. E: as in D but at 0.1mM external [Ca2+]o. F: percentage of cells that oscillate at 0.1mM [Ca2+]o with respect to cells that oscillate at 1.5mM [Ca2+]o. RyR2R420Q vs RyR2WT. aP<.05. bP<.01.](https://static.elsevier.es/multimedia/18855857/0000006800000005/v1_201504231305/S1885585714003314/v1_201504231305/en/main.assets/gr6.jpeg?xkr=eyJpdiI6IktuUndZUnVqSkVzb05EUU1ieEFjbkE9PSIsInZhbHVlIjoiazI3ZWFDcDZDOURyNk1JZWVNa2FMa2pqNWswakUySldmTkdaQXUySUlMRzdwK3Z3Y1ZMZUNuV2ZSTDV3UHFLZEJHclZ1T3VIdGN0bWRZdUVvamVjb1VLR2ZNeVZFWGVJOHN1M2hkVUVyZUtzL1pFTFVHUCsvenBHc1h6dWZxaWp1NzNINE00cWVvRzhiMWowK3pqVWh2R0p1cXdneWtxU2NSUTBabEVDWS9pWkgvNGNLb0hTTUthUWRUTGhKNnowbjFxcEtrQ0tjTEZSbWhYM3M3TGx5eHowKzVEeG0zckc3b0c3bzRDcUpub3NGdk9tTktNZnZLNWYwVERjVjRvSy9oVlVTNnovc0ZmUGdKTk1wY1pJNUcvSy91eUNyakdjanlURDBQL3ZIZWM9IiwibWFjIjoiMjZmZDM3ZDdkNDM0NjU0YmFiODA0ZmE3MTBiYjIzYzJjYWZjNTMzY2M5ODRhMTIxNmI3M2ZlMTljYTg0MDRkOSIsInRhZyI6IiJ9 "Spontaneous Ca2+ oscillations in HEK-293 cells expressing RyR2R420Q are smaller and slower but change to faster than RyR2WT at lower [Ca2+]o. A: time series of confocal images in intact HEK-293 cells expressing RyR2WT (left) and RyR2R420Q (right). B: representative fluorescence profiles in intact HEK-293 cells expressing RyR2WT (left) and RyR2R420Q (right), at 1.5mM [Ca2+]o. C: amplitude of Ca2+ oscillations at 1.5mM [Ca2+]o. D: as in C but for the cycle length between consecutive Ca2+ oscillations. E: as in D but at 0.1mM external [Ca2+]o. F: percentage of cells that oscillate at 0.1mM [Ca2+]o with respect to cells that oscillate at 1.5mM [Ca2+]o. RyR2R420Q vs RyR2WT. aP<.05. bP<.01.")
Spontaneous Ca2+ oscillations in HEK-293 cells expressing RyR2R420Q are smaller and slower but change to faster than RyR2WT at lower [Ca2+]o. A: time series of confocal images in intact HEK-293 cells expressing RyR2WT (left) and RyR2R420Q (right). B: representative fluorescence profiles in intact HEK-293 cells expressing RyR2WT (left) and RyR2R420Q (right), at 1.5mM [Ca2+]o. C: amplitude of Ca2+ oscillations at 1.5mM [Ca2+]o. D: as in C but for the cycle length between consecutive Ca2+ oscillations. E: as in D but at 0.1mM external [Ca2+]o. F: percentage of cells that oscillate at 0.1mM [Ca2+]o with respect to cells that oscillate at 1.5mM [Ca2+]o. RyR2R420Q vs RyR2WT. aP<.05. bP<.01.
The present report on a highly symptomatic CPVT family describes for the first time the phenotype associated with the RyR2R420Q mutation, highlighting a high incidence of non-VA. Our in vitro results confirm a dysfunction of the mutant channel.
The global yield of the initial ET for CPVT diagnosis (sensitivity, 89%; positive predictive value, 100%, and negative predictive value, 93%) was higher than that reported in other series.26 The behavior of two young genotype+ patients (IV:4 and IV:8) suggests that penetrance could be age-related. Additionally, sinus bradycardia, atrial and/or junctional tachyarrhythmias and postexercise giant U-waves were present in RyR2R420Q carriers (Figures 3 and 4). U-wave alternans and post-extrasystolic U-wave polarity changes27 (as a result of an altered Ca2+cycling), and also increased U/T28,29 (regarded as the electrocardiographic counterpart of delayed after depolarizations), have occasionally been described in CPVT individuals. Of note, our CPVT patients exhibited post-extrasystolic U-wave augmentation and increased U/T on exertion, precisely under catecholaminergic drive. The origin of supraventricular arrhythmias could also be ascribed to the RyR2 mutation, as RyR2 is expressed in all cardiomyocytes, including pacemaker cells and atrial myocytes.30,31 In fact, we recently found that the RyR2R4496C mutation promotes an increased automatism in ventricular cells15 but a decreased automatism in sinoatrial nodal cells.9 Thus, mutated RyR2 channels appear to exhibit cell–type-dependent dysfunctional activity.
The RyR2R420Q belongs to the N-terminal cluster and provokes the substitution of the most strongly charged basic amino acid (arginine) by a polar uncharged residue (glutamine). This mutation, recently reported in 4 unrelated patients, lacks clinical and functional characterization.14,32 Remarkably, the residue 420 is highly conserved across species32 and behaves as a hot spot, given that different substitutions have been described at the same point and near it (R420W3,33-35 and I419F36,37). Notably, the RyR2R420Q shows a higher penetrance than the RyR2R420W (91% vs 25%), and does not display any feature of arrhythmogenic cardiomyopathy.33,34
So far, most CPVT mutations in which functional studies have been performed behave as gain-of-function.35,38 Increased Ca2+ sensitivity (luminal or cytosolic),15,24,38,39 an altered interdomain interaction between the N-terminal and central domains,40,41 and diminished FKBP12.6 binding38,39 account for the most accepted mechanisms. Very recently, a disrupting effect of the RyR2R420Q has been observed in the crystal structure of the N-terminal region, since it ablates chloride binding, disturbing its domain folding42,43 even though, in linear terms, the 420 residue is very far from the proposed molecular region involved in Ca2+-dependent activation (residues 4485-4494).40 The N-terminus has also been shown to be an important structural region that has the ability to self-tetramerise, and may be involved in regulation of native channel function.44 Thus, it appears clear from a static structural standpoint that the RyR2R420Q destabilizes the inter-subunit interface, probably facilitating the channel opening.42-45
Herein, we offer the first initial assessment of the RyR2R420Q mutation from a functional perspective. First, our findings clearly reflect that the RyR2R420Q mutation behaves as a gain-of-function both in permeabilized and intact cells (Figures 5D, 5F, 6E and 6F) at very low [Ca2+]i, similar to that measured in rat and human cardiomyocytes.46,47 Second, the reason for the reduced peak of caffeine-evoked [Ca2+] release in RyR2R420Q cells (Figure 5E) does not have a straightforward interpretation. It could be related to a hypoactive channel at higher cytosolic Ca2+ but without significant shift in EC50. Finally, in intact RyR2R420Q cells, spontanous oscillations had decreased amplitude and frequency (Figures 6B and 6D). This decrease could be reminiscent of the sinus bradycardia found in CPVT patients (Figure 3B), and it is not dependent of lower sarcoplasmic reticulum Ca2+ load, since this was normal (see “Results”).
Study LimitationsHeterologous cell expression systems are useful to characterize RyR2 mutations, although they may present limitations because they cannot replicate the electrical behavior in vivo, where other proteins and the autonomic tone may interact with the dysfunctional channel. Thus, experiments in either transgenic mice or iPS-CM (induced pluripotent stem cell-derived cardiomyocytes) will provide more definitive conclusions.
CONCLUSIONSThis work provides a novel and detailed clinical characterization of the RyR2R420Q mutation. Our analyses highlight that non-VA are common and might reinforce the suspicion of CPVT in an appropriate scenario. Furthermore, in vitro analyses suggest that the RyR2R420Q results in an aberrant channel.
FUNDINGThis work was supported by the Instituto de Salud Carlos III (PI14/01477, RD12/0042/0029), Sociedad Española de Cardiología (Proyecto de Investigación Clínica en Cardiología Dr. Pedro Zarco), Biobanco La Fe (PT13/0010/0026), Prometeo 2011/027, ANR (Agence Nationale de la Recherche) (ANR-13-BSV1-0023), and Région Île-de-France (CORDIM, COD 100256). INSERM (Institut National de la Santé et de la Recherche Médicale) U-769 is a member of the LERMIT (Laboratory of Excellence in Research on Medication and Innovative Therapeutics), supported by a grant ANR Investissements d’avenir. Patricia Neco was supported by Fondation pour la Recherche Médicale and Spyros Zissimopoulos, by the British Heart Foundation.
CONFLICTS OF INTERESTNone declared.