In clinical practice in the context of early disease detection and prevention of common adult-onset conditions, one of the most common problems is classification or decision-making, which is carried out through diagnostic and/or prognostic tests. When seeking their optimization, it is fundamental to be aware of their accuracy and precision.
Since the completion of the Human Genome Project, the combination of large-scale genome variation projects, such as the HapMap and 1000 Genomes projects, together with low-cost robust genotyping platforms and the rapid advance of DNA sequencing technologies, has enabled genome-wide association studies (GWAS) in large cohorts and exome- and genome-wide sequencing studies. Consequently, there has been an exponential increase in the abundance of individual-specific genotype data, leading to the era of personalized medicine or precision genomics-based medicine.1
Historically, genetic diseases were classified into those with a Mendelian or simple inheritance caused by genetic variations with a large effect and those with a complex inheritance caused by the sum of genetic variations with a reduced effect. However, currently, each individual's overall risk of developing a common disease is probably marked by a combination of common low-risk genetic variants and rare high-risk genetic variants.2
GWAS have focused on identifying disease- or trait-associated genetic variants (typically single nucleotide polymorphisms [SNPs]), which are common in a given population (eg, minor allele frequency > 1%). To date, GWAS have identified thousands of loci that are associated with several complex human traits and diseases, including cardiovascular diseases.3 Notably, many of the loci previously associated with these complex human diseases are highlighted by multiple low-risk SNPs.4
These data have provided numerous insights into the genes and pathways that cause disease, but more recently there has been increasing interest in the use of these data for disease risk prediction.5,6 In the last decade, genomics-based precision medicine has consistently emerged to provide effective and tailored health care for patients, depending on their genetic background. The inclusion of genetic risk scores (GRS), including disease or phenotype associated SNPs, into risk modelling has improved the accuracy of individual disease prediction,7 as reported in an original article published by Rincón et al. in Revista Española de Cardiología.8
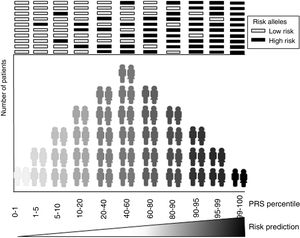
The main focus of the development of genetic risk models is to achieve accurate predictive power for recognizing at-risk individuals (figure 1). Most commonly these models are calculated as a weighted sum of the number of risk alleles carried by an individual, where the risk alleles and their effect sizes are defined by previous GWAS.6 Therefore, the accuracy of a GRS is marked by the efficiency of previous GWAS studies in finding genetic variants associated with common diseases. In other words, the sounder the foundations of the building—in our case the genetic associations described in the GWAS studies—the more resistant our construction will be, ie, the more accurate our risk prediction estimate will be.

Predictive performance is typically evaluated by receiver operating characteristic (ROC) curves, in which the sensitivity and specificity of the predictions are ranked at various cutoff values. In the simplistic case, in which the development of any condition or disease is to be predicted, sensitivity is given as the fraction of the true-positive ratio among the total number of patients with the disease. Of note, a true positive is any patient who has the disease and has a positive result in the clinical prediction model. Therefore, the true-positive ratio is the probability of correctly classifying a patient. The specificity is the true-negative ratio among the total number of patients without the disease. Specificity is the probability of correctly classifying a healthy individual, that is to say, the probability that a healthy person will have a negative result. A ROC curve is a 2-dimensional graph in which the true-positive rate (sensitivity) is represented on the vertical axis, and the false-positive rate (1-specificity) on the horizontal axis. Therefore, a ROC graph of a prediction model represents the relative equilibrium between true positives and false negatives. The area under the ROC curve is the probability of the examined model correctly identifying a case out of a randomly chosen pair of case and control samples. Area under ROC curve results range from 0.5 (ie, random) to 1 (ie, 100% accuracy).
GRS or polygenic risk scores (PRS), as many authors now call them, rather than predict the presence or absence of a disease, aim to classify the population into different risk levels (figure 1). The threshold for considering a positive GRS depends on the balance between the risk value marked by their own cutoff values in combination with other risk factors and the benefits associated with a possible therapeutic or lifestyle intervention.
It is currently believed that the genetics of nonfamilial forms of the most common adult-onset heart diseases are mainly linked to a combination of common variants with small effect sizes distributed throughout the genome and rare variants of moderate effect in genes known to cause familial disease. Evidence of this has been described in recent comprehensive genomic studies, such as an extensive GWAS coronary artery disease study9 and a large-scale sequencing study of type 2 diabetes mellitus.10 Therefore, the effect of each of these common variants on an individual will be too small to predict risk, but the combination of many of these common variants can be used to predict risk efficiently, especially if risk is predicted in combination with classic risk factors, such as clinical risk factors or certain environmental exposures.
One of the first publications on the implementation of GRS in cardiovascular diseases was the study by Morrison et al.,11 in which the use of an 11-polymorphism score for predicting coronary heart disease risk did not improve the predictive capacity of classic risk factors. Since then, many PRS have been published and validated and are especially effective in groups of patients with highly specific phenotype. Some examples are coronary artery disease PRS aiming to individualize the decision to initiate lifetime statin therapy,12 or PRS to improve the prediction capacity of patients classified as being at intermediate risk of cardiovascular heart disease according to the Framingham scale.13
GRS applied to young adults to predict recurrent events after myocardial infarction, as described by Rincón et al.,8 should be validated in a more extensive sample, since positive results are mainly seen in young patients without diabetes, but the observation is based on a small number of patients. Studies like this one open doors to the implementation of PRS in clinical prediction models but they must always be validated and based on extensive data.
Last but not least, we must not forget the uncertainty in the estimation of the effect size associated with each common variant included in a genetic score, when the PRS is used to estimate the risk in other populations beyond the population studied in the GWAS. Since most of the GWAS were executed in European ancestry populations and genetic diversity among populations with different ancestry is well known, we must take special care when extending the applicability of PRS to all populations worldwide. Estimates are not transferable between populations, and ultimately the PRS is applied to an individual patient with a given geographical origin and with a characteristic genetic load, but with the same rights to health care.14
Although the number of studies with polygenic risk estimates has grown exponentially in the last 5 years, large-scale studies should be carried out to demonstrate the usefulness of polygenic risk estimation, not only in the cardiovascular field but also in other areas of human health. In this regard, the European action of the One Million Genomes Initiative, aiming to have this number of genomes linked to clinical data sequenced for 2022, with Spain a Signatory Member State, is perhaps the most promising project.15 Whole-genome data at this scale have the potential to make rapid progress in precision medicine and risk prediction estimates.
FundingThis work was partially suported by Plan Estatal de I+D+i 2013-2016, Subdirección General de Evaluación y Fomento de la Investigación (ISCIII-SGEFI) from Instituto de Salud Carlos III (ISCIII) and Fondo Europeo de Desarrollo Regional (FEDER) (grant numbers PI16/00903, CB16/11/00226, CB06/07/0088).
Conflicts of interestNone declared.
.