


First generation drug-eluting stents have considerably reduced in-stent restenosis and broadened the applications of percutaneous coronary interventions for the treatment of coronary artery disease. The polymer is an integral part of drug-eluting stents in that, it controls the release of an antiproliferative drug. The main safety concern of first generation drug-eluting stents with permanent polymers—stent thrombosis—has been caused by local hypersensitivity, delayed vessel healing, and endothelial dysfunction. This has prompted the development of newer generation drug-eluting stents with biodegradable polymers or even polymer-free drug-eluting stents. Recent clinical trials have shown the safety and efficacy of drug-eluting stents with biodegradable polymer, with proven reductions in very late stent thrombosis as compared to first generation drug-eluting stents. However, the concept of using a permanent metallic prosthesis implies major drawbacks, such as the presence of a foreign material within the native coronary artery that causes vascular inflammation and neoatherosclerosis, and also impedes the restoration of the vasomotor function of the stented segment. Bioresorbable scaffolds have been introduced to overcome these limitations, since they provide temporary scaffolding and then disappear, liberating the treated vessel from its cage. This update article presents the current status of these new technologies and highlights their future perspectives in interventional cardiology.
Keywords
Coronary stents were first developed in the mid 1980s to overcome the inherent limitations of balloon angioplasty, including elastic recoil and vessel closure in the acute phase, as well as constrictive remodeling and restenosis in the late phase.1–3 In the 1990s, this technology became widely accepted as a promising treatment strategy for patients with coronary artery disease after the landmark Belgian Netherlands Stent trial, which demonstrated the superiority of the bare metal stent (BMS) over balloon angioplasty.4 Although coronary stenting improved angiographic results and clinical outcomes, neointimal hyperplasia and restenosis continued to be major limitations of this technology.5 In order to minimize neointimal hyperplasia and thereby reduce repeat revascularization, drug-eluting stents (DES) were developed. Early pivotal trials of the first generation DES showed excellent results with respect to the reduction of in-stent restenosis, such that they rapidly replaced BMS.6,7 In the year 2006, safety concerns were raised with DES following reports linking their use to an increased risk of stent thrombosis (ST).8,9 First generation DES, with permanent polymers, have been associated with delayed endothelialization, endothelial dysfunction, and local hypersensitivity reactions, resulting in an increased risk of ST and the need for prolongation of dual antiplatelet therapy.10,11
Newer generation DES, with thinner struts and more biocompatible polymers, have considerably improved their safety profile.12–15 However, concerns still persist over the presence of durable polymers, as evidence from animal and human studies still suggest that these durable polymers may cause persistent arterial wall inflammation and delayed vascular healing, both of which may subsequently have a potential role in precipitating ST and delayed in-stent restenosis (ie, late catch-up phenomenon).16 Newer generation DES, coated with biodegradable polymers, offer the attractive combination of controlled drug elution in parallel with biodegradation of the polymer into inert monomers. After the completion of biodegradation, only a “BMS” remains, thereby reducing the long-term risks associated with the presence of a permanent polymer.17 An extension of this concept has brought the development of newer DES that are completely free of polymer, or come with novel coatings. In addition, bioresorbable metallic (ie, magnesium) and polymeric scaffolds have been developed, which initially safeguard the patency of the treated vessel and then disappear. The aim of this article is to review new stent technologies that are currently undergoing clinical investigation and discuss their future perspectives in interventional cardiology.
NEW GENERATION METALLIC DRUG ELUTING STENTDrug Eluting Stent With Biodegradable PolymersBiodegradable polymeric coatings facilitate drug delivery to the vessel wall and are then resorbed without any long-term sequelae. Since their introduction in the year 2004,18 many DES with biodegradable polymers have been developed, particularly after it was hypothesized that this technology would potentially reduce the risk of very late ST (VLST), an adverse event which has been associated with durable-polymer DES. The randomized ISAR-TEST 4 trial was conducted to test the noninferiority of a biodegradable-polymer rapamycin-eluting stent (RES; Yukon Choice PC, Translumina; Hechingen, Germany) to a durable-polymer DES (ie, the first generation Cypher sirolimus-eluting stent [SES] or the second generation Xience V everolimus-eluting stent [EES]), with respect to clinical outcomes. A total of 2603 patients were enrolled in the trial. At 3-year follow-up, there were no significant differences in a composite of cardiac death, target-vessel myocardial infarction (MI), and target lesion revascularization (TLR) (RES 20.1% vs DES 20.9%, P=.59), as well as the incidence of definite/probable ST (RES 1.4% vs DES 1.9%, P=.51).19 Longer-term clinical follow-up is required to evaluate the potential superiority of RES over the traditional DES in reducing the risk of VLST.
Biolimus-eluting Stent With Biodegradable PolymerBiolimus A9 is a semisynthetic limus-drug designed for stent application which has a similar potency to sirolimus, but is 10 times more lipophilic. It is immersed at a concentration of 15.6 µg/mm into a polylactic acid biodegradable polymer that covers the abluminal stent surface. Polylactic acid is coreleased with biolimus and completely metabolized into carbon dioxide and water over 6 months to 9 months. The stainless steel stent platform has a strut thickness of 112 µm, with a quadrature link design. Currently, the stent platforms utilizing this technology are the BioMatrix Flex (Biosensors Inc.; Singapore), NOBORI (Terumo Corp.; Tokyo, Japan), and Axxess (Biosensors Inc.).
In the LEADERS trial, the BioMatrix stent was shown to be noninferior to the first generation durable-polymer Cypher SES, with respect to a composite end point of cardiac death, MI, and ischemia-driven target vessel revascularization at 12-month follow-up (BioMatrix 10.6% vs Cypher 12.0%, P=.37).20 This noninferiority has recently been confirmed at 5-year follow-up.21 Importantly, the BioMatrix stent showed a significantly lower incidence of definite VLST at 5-year follow-up (hazard ratio=0.26 [0.10-0.68]). A pooled data analysis of the randomized ISAR-TEST 3, ISAR-TEST 4, and LEADERS trials also showed that the DES with biodegradable polymers were associated with a lower risk of VLST as well as MI compared to the Cypher SES.22 The LEADERS trial not only provided the first evidence of improved clinical outcomes compared to the first generation DES, but is also the proof of concept in terms of biodegradable-polymer DES.
Everolimus-eluting Stent With Biodegradable Polymer: SYNERGY StentThe SYNERGY stent (Boston Scientific; Natick, Massachusetts, United States) consists of a thin-strut (74µm), platinum-chromium platform that delivers everolimus from a bioabsorbable poly-lactide-co-glycolide polymer applied to the abluminal surface. In the randomized, EVOLVE trial, the safety and efficacy of 2 dose formulations (standard dose [SD], 113µg/20mm, and half dose [HD], 56µg/20mm) of the SYNERGY stent were compared to the durable-polymer PROMUS Element EES (Boston Scientific).23 A total of 291 patients were randomly assigned in a 1:1:1 ratio to SYNERGY, SYNERGY HD, and EES. The primary clinical endpoint was the 30-day rate of target lesion failure (TLF), defined as a composite of cardiac death, MI related to the target vessel, and TLR. TLF occurred in 3.1%, 1.1%, and 0% of patients in the SYNERGY, SYNERGY HD, and EES groups, respectively. The 6-month in-stent late lumen loss (LLL) was 0.10mm for SYNERGY, 0.13mm for SYNERGY HD, and 0.15mm for EES (Pnoninferiority<.001). There were no ST events in any group at up to 6-month follow-up. Recently, the SYNERGY stent acquired the Conformité Européenne (CE) mark approval; a pivotal EVOLVE II trial aiming a head-to-head comparison of 12-month TLF with SYNERGY (842 patients) and EES (842 patients) is currently ongoing.
Other Drug Eluting Stents With Biodegradable PolymersCurrently, many DES with biodegradable polymers are commercially available or under clinical investigation (Table 1). Preliminary studies have shown comparable results at 6 months to 9 months to that of aforementioned DES with biodegradable polymers. Although biodegradable polymers appear to have become a promising drug-delivery technology in the newer generation DES platform, there are issues remaining to be addressed before their widespread clinical application.24 Further research is needed in order to optimize the composition and release kinetics of these polymers.
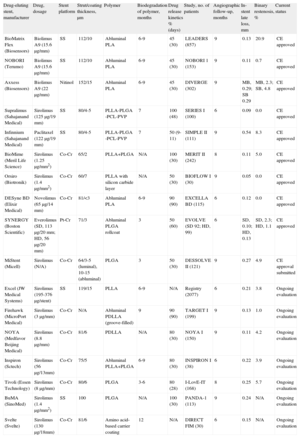
Overview of Drug-eluting Stents With a Biodegradable Polymer Under Clinical Investigation or Already Available Outside the United States
| Drug-eluting stent, manufacturer | Drug, dosage | Stent platform | Strut/coating thickness, µm | Polymer | Biodegradation of polymer, months | Drug release kinetics % (days) | Study, no. of patients | Angiographic follow-up, months | In-stent late loss, mm | Binary restenosis, % | Current status |
| BioMatrix Flex (Biosensors) | Biolimus A9 (15.6µg/mm) | SS | 112/10 | Abluminal PLA | 6-9 | 45 (30) | LEADERS (857) | 9 | 0.13 | 20.9 | CE approved |
| NOBORI (Terumo) | Biolimus A9 (15.6µg/mm) | SS | 112/10 | Abluminal PLA | 6-9 | 45 (30) | NOBORI 1 (153) | 9 | 0.11 | 0.7 | CE approved |
| Axxess (Biosensors) | Biolimus A9 (22µg/mm) | Nitinol | 152/15 | Abluminal PLA | 6-9 | 45 (30) | DIVERGE (302) | 9 | MB, 0.29; SB 0.29 | MB, 2.3; SB, 4.8 | CE approved |
| Supralimus (Sahajanand Medical) | Sirolimus (125µg/19 mm) | SS | 80/4-5 | PLLA-PLGA -PCL-PVP | 7 | 100 (48) | SERIES I (100) | 6 | 0.09 | 0.0 | CE approved |
| Infinnium (Sahajanand Medical) | Paclitaxel (122µg/19 mm) | SS | 80/4-5 | PLLA-PLGA -PCL-PVP | 7 | 50 (9-11) | SIMPLE II (111) | 9 | 0.54 | 8.3 | CE approved |
| BioMime (Meril Life Science) | Sirolimus (1.25µg/mm2) | Co-Cr | 65/2 | PLLA+PLGA | N/A | 100 (30) | MERIT II (242) | 8 | 0.11 | 5.0 | CE approved |
| Orsiro (Biotronik) | Sirolimus (1.4µg/mm2) | Co-Cr | 60/7 | PLLA with silicon carbide layer | N/A | 50 (30) | BIOFLOW I (30) | 9 | 0.05 | 0.0 | CE approved |
| DESyne BD (Elixir Medical) | Novolimus (65µg/14 mm) | Co-Cr | 81/<3 | Abluminal PLA | 6-9 | 90 (90) | EXCELLA BD (115) | 6 | 0.12 | 0.0 | CE approved |
| SYNERGY (Boston Scientific) | Everolimus (SD, 113µg/20 mm; HD, 56µg/20 mm) | Pt-Cr | 71/3 | Abluminal PLGA rollcoat | 3 | 50 (60) | EVOLVE (SD 92; HD, 99) | 6 | SD, 0.10; HD, 0.13 | SD, 2.3; HD, 1.1 | CE approved |
| MiStent (Micell) | Sirolimus (N/A) | Co-Cr | 64/3-5 (luminal), 10-15 (abluminal) | PLGA | 3 | 50 (30) | DESSOLVE II (121) | 9 | 0.27 | 4.9 | CE approval submitted |
| Excel (JW Medical Systems) | Sirolimus (195-376µg/stent) | SS | 119/15 | PLLA | 6-9 | N/A | Registry (2077) | 6 | 0.21 | 3.8 | Ongoing evaluation |
| Firehawk (MicroPort Medical) | Sirolimus (3µg/mm) | Co-Cr | N/A | Abluminal PDLLA (groove-filled) | 9 | 90 (90) | TARGET I (199) | 9 | 0.13 | 1.0 | Ongoing evaluation |
| NOYA (Medfavor Beijing Medical) | Sirolimus (8.8µg/mm) | Co-Cr | 81/6 | PDLLA | N/A | 80 (30) | NOYA I (150) | 9 | 0.11 | 4.2 | Ongoing evaluation |
| Inspiron (Sctech) | Sirolimus (56µg/13mm) | Co-Cr | 75/5 | Abluminal PLLA+PLGA | 6-9 | 80 (30) | INSPIRON I (38) | 6 | 0.22 | 3.9 | Ongoing evaluation |
| Tivoli (Essen Technology) | Sirolimus (8µg/mm) | Co-Cr | 80/6 | PLGA | 3-6 | 80 (28) | I-LovE-IT (168) | 8 | 0.25 | 5.7 | Ongoing evaluation |
| BuMA (SinoMed) | Sirolimus (1.4µg/mm2) | SS | 100 | PLGA | N/A | 100 (30) | PANDA-1 (113) | 9 | 0.24 | N/A | Ongoing evaluation |
| Svelte (Svelte) | Sirolimus (130µg/18mm) | Co-Cr | 81/6 | Amino acid-based carrier coating | 12 | N/A | DIRECT FIM (30) | 6 | 0.15 | N/A | Ongoing evaluation |
CE, Conformité Européenne; Co-Cr, cobalt-chromium; HD, half dose; MB, main branch; N/A, not applicable; PCL, poly-L-lactide-co-e-caprolactone; PDLLA, poly-D, L-lactic acid; PLA, polylactic acid; PLGA, poly-lactide-co-glycolide; PLLA, poly-L-lactic acid; Pt-Cr, platinum-chromium; PVP, poly-vinyl-pyrrolidone; SB, side branch; SD, standard dose; SS, stainless steel.
The next major step forward may be metallic stent structures which allow for appropriate drug-elution kinetics without the use of polymers. Several devices have been designed to test this approach by incorporating drugs into a microporous or nanoporous surface of the stent (Table 2). The efficacy of a polymer-free SES (SES-PF; Yukon Choice, Translumina) was investigated in the ISAR-TEST 3 trial.25,26 The SES-PF (201 patients) was compared to a SES with a biodegradable polymer (202 patients, SES-BP; Yukon Coice PC, Translumina) and a SES with a permanent polymer (202 patients, SES-PP; Cypher, Cordis; Miami Lakes, Florida, United States). At 2 years, there were no significant differences in death, MI (SES-PF 6.5%, SES-BP 5.9%, and SES-PP 6.4%), TLR (SES-PF 13.4%, SES-BP 8.4%, and SES-PP 10.4%), and definite/probable ST (SES-PF 1.0%, SES-BP 0.5%, and SES-PP 1.0%). Patients undergoing paired angiography at 6 months to 8 months and at 2 years (302 patients), demonstrated a lower delayed LLL in the SES-PF (-0.01 mm) group. as compared to both the SES-BP (0.17mm) and the SES-PP (0.16 mm) (P<.001) groups. The absence of delayed LLL in the SES-PF group may indicate a lower propensity for stent-vessel wall interactions, owing to less inflammatory or hypersensitive reactions. Recently, the 5-year clinical outcomes in the ISAR-TEST trial have been reported.27 There were no statistically significant differences in ST events between SES-PF and the first generation TAXUS paclitaxel-eluting stent (PES) (SES-PF 0.5% vs PES 1.6%, P=.32). Extended follow-up data may further support the durability, safety, and efficacy of the SES-PF.
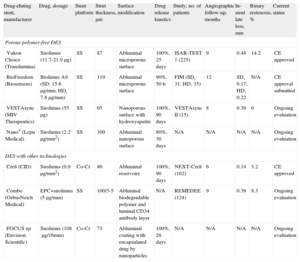
Overview of Polymer-free and Novel-coating Drug-eluting Stents Under Clinical Investigation or Already Available Outside the United States
| Drug-eluting stent, manufacturer | Drug, dosage | Stent platform | Strut thickness, µm | Surface modification | Drug release kinetics | Study, no. of patients | Angiographic follow-up, months | In-stent late loss, mm | Binary restenosis, % | Current status |
| Porous polymer-free DES | ||||||||||
| Yukon Choice (Translumina) | Sirolimus (11.7-21.9µg) | SS | 87 | Abluminal microporous surface | 100%, 25 days | ISAR-TEST 1 (225) | 9 | 0.48 | 14.2 | CE approved |
| BioFreedom (Biosensors) | Biolimus A9 (SD, 15.6µg/mm; HD, 7.8µg/mm) | SS | 119 | Abluminal microporous surface | 90%, 50 h | FIM (SD, 31; HD, 35) | 12 | SD, 0.17; HD, 0.22 | N/A | CE approval submitted |
| VESTAsync (MIV Therapeutics) | Sirolimus (55µg) | SS | 65 | Nanoporous surface with hydroxyapatite | 100%, 90 days | VESTAsync II (15) | 8 | 0.39 | 0 | Ongoing evaluation |
| Nano+ (Lepu Medical) | Sirolimus (2.2µg/mm2) | SS | 100 | Abluminal nanoporous surface | 80%, 30 days | N/A | N/A | N/A | N/A | Ongoing evaluation |
| DES with other technologies | ||||||||||
| Cre8 (CID) | Sirolimus (0.9µg/mm2) | Co-Cr | 80 | Abluminal reservoirs | 100%, 90 days | NEXT-Cre8 (162) | 6 | 0.14 | 3.2 | CE approved |
| Combo (OrbusNeich Medical) | EPC+sirolimus (5µg/mm) | SS | 100/3-5 | Abliminal biodegradable polymer and luminal CD34 antibody layer | N/A | REMEDEE (124) | 9 | 0.39 | 8.3 | Ongoing evaluation |
| FOCUS np (Envision Scientific) | Sirolimus (108µg/16mm) | Co-Cr | 73 | Abluminal coating with encapsulated drug by nanoparticles | 100%, 28 days | N/A | N/A | N/A | N/A | Ongoing evaluation |
CE, Conformité Européenne; Co-Cr, cobalt-chromium; SD, standard dose; DES, drug-eluting stent; EPC, endotheloal progenitor cell (capture technology); HD, half dose; N/A, not applicable; SS, stainless steel.
The BioFreedom stent (Biosensors Inc.). is a 316L stainless steel, polymer-free stent that is coated with biolimus A9 (Fig. 1). Preclinical studies have reported lower injury scores; lower numbers of struts with fibrin, granulomas, and giant cells; significantly lower percentage of diameter stenosis; and greater endothelialization with the BioFreedom stent at 180-day follow-up as compared to the Cypher SES.28 The first-in-man (FIM) trial enrolled 182 patients who were randomized to receive either BioFreedom with SD sirolimus (15.6µg/mm), BioFreedom with low dose (LD) sirolimus (7.8µg/mm), or TAXUS Liberté PES. At 12 months, the in-stent LLL was 0.17mm in the BioFreedom SD arm (P<.0001 vs PES), 0.22mm in the BioFreedom LD arm (P=.21 vs PES), and 0.35mm in the PES arm. There were no ST events and no differences in major adverse cardiac events (MACE) including all-cause death, MI, and emergent bypass surgery or TLR at up to 36 months (BioFreedom SD 11.9%; BioFreedom LD 18.1%, and PES 10.0%).29 Currently, a randomized LEADERS FREE trial has been planned to examine the noninferiority (a composite of cardiac death, MI, and ST) and superiority (clinically driven TLR) of the BioFreedom stent to BMS in >2400 elderly patients with dual antiplatelet therapy for 1 month after stent implantation.
VESTAsync Stent
The VESTAsync stent (MIV Therapeutics; Atlanta, Georgia, United States) combines a stainless steel platform with a nanoporous, hydroxyapatite (biocompatible crystalline derivative of calcium phosphate) surface coating that is impregnated with 55µg of sirolimus mixture (Fig. 2). It is expected that sirolimus will be completely released within the first 3 months after the implantation, and that the hydroxyapatite will be stable over 4 months. The safety and efficacy of the VESTAsync stent was evaluated in the VESTAsync I FIM trial. A total of 15 patients with single de novo coronary artery lesions were enrolled. In-stent LLL was 0.36mm at 9 months, with no MACE reported at up to 1-year follow-up.30 Recently a randomized VESTAsync II trial has been reported.31 The patients treated with the VESTAsync stent (50 patients) showed a significantly lower in-stent LLL compared to those treated with BMS (25 patients) at 8 months (VESTAsync 0.39mm vs BMS 0.74mm, P=.03). No evidence of ST was reported at up to 2-year follow-up.
, cross section of the hydroxyapatite coating (B), final coating including the hydroxyapatite filled with sirolimus formulation (C), and cross section of the final coating (D). A schematic representation of the surface coating (E).")
The polymer-free VESTAsync sirolimus-eluting stent system. The scanning electron microscopy images of microporous hydroxyapatite coating (A), cross section of the hydroxyapatite coating (B), final coating including the hydroxyapatite filled with sirolimus formulation (C), and cross section of the final coating (D). A schematic representation of the surface coating (E).
The Nano+ stent (Lepu Medical; Beijing, China) is a stainless steel, polymer-free stent with a nanoporus surface coated with sirolimus (2.2µg/mm2). The average diameter of the pores is approximately 400nm and almost 80% of the drug is programmed to be released within 30 days. The Bicare stent is another nanopore-based polymer-free DES, which uses both sirolimus and probucol. These 2 stents have a similar design, the only difference between the Bicare stent and the Nano+ stent being the drug type. Thirty patients with de novo lesions were enrolled in the Bicare FIM trial.32 In-stent LLL was 0.14mm and the rate of tissue coverage of the struts was 98.3% determined by optical coherence tomography (OCT) at 4 months.32 Similarly, the ISAR-TEST 5 trial demonstrated the noninferiority of polymer-free sirolimus and probucol-eluting stents (Yukon Choice, Translumina) over the second generation durable-polymer resolute zotarolimus-eluting stents (Medtronic Cardiovascular; Santa Clara, California, United States) in terms of MACE and ST at 1-year follow-up.33 A post-market study of the Nano+ stent is currently ongoing in China, and another trial aiming the CE mark approval has been planned in Europe.
Drug Eluting Stent With Other TechnologiesCre8 StentThe Cre8 stent (CID; Saluggia, Italy) is a polymer-free stent that is integrally coated with an ultra-thin (0.3µm) passive carbon coating (i-Carbofilm, CID). The amphilimus formulation, constituted by sirolimus (0.9µg/mm2) with an excipient composed of a long-chain fatty acid mixture to modulate the drug release, is loaded into abluminal reservoirs. Complete release of sirolimus is expected within the first 3 months after stent deployment. A total of 323 patients were randomized to receive either Cre8 (162 patients) or TAXUS Liberté PES (161 patients) in the NEXT FIM trial.34 The primary endpoint was in-stent LLL at 6 months, and was significantly lower in the Cre8 group (Cre8 0.14mm vs PES 0.34mm, P<.0001). A cumulative incidence of MACE including cardiac death, MI, and TLR in the Cre8 group was 6.7% at 2 years, showing no differences compared to PES (7.1%). Only 1 case of definite late ST was observed in each group at up to 2-year follow-up.35 An all-comers registry (1000 patients) is currently ongoing and is expected to complete enrollment of patients by early 2013.
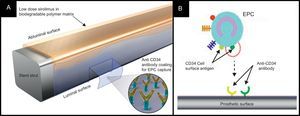
Combo StentThe Combo stent (OrbusNeich Medical; Hong Kong, China) applies the endothelial progenitor cell capture technology to enhance vessel healing (ie, immobile CD34 antibodies on the luminal surface of the strut), incorporating abluminal LD sirolimus and a biodegradable polymer into the current DES technology (Fig. 3). Data from OCT and histology at 28 days in a porcine model indicated that this hybrid stent promotes endothelialization, and reduces neointimal formation and inflammation when compared to the Cypher SES and the first generation Genous endothelial progenitor cell stent.36 The REMEDEE FIM trial randomized 180 patients to treatment with either the Combo stent (124 patients) or the TAXUS Liberté PES (59 patients). The in-stent LLL at 9 months was 0.39mm in the Combo group and 0.44mm in the PES group (Pnoninferiority=.0012). Binary restenosis was observed in 8.3% patients in the Combo group and in 13.5% patients in the PES group (P=.30). No cases of ST were reported in both groups at up to 9-months follow-up.37 Further investigation is required to determine the prohealing effect and clinical efficacy of this device.

The Combo dual therapy stent system. A: The Combo stent consists of an abluminal biodegradable polymer matrix with a sirolimus and a luminal CD-34 antibody layer. B: A schematic representation of the endothelial progenitor cells capture technology. The CD-34 antigens on the surface of the endothelial progenitor cells attach to the anti-CD-34 antibodies on the stent's surface, promoting endothelialization. EPC, endothelial progenitor cells.
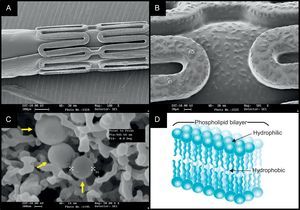
The FOCUS np stent (Envision; Surat, India) platform has a novel carrier; a phospholipid 2-layer nanoparticle that encapsulates sirolimus (Fig. 4). The encapsulated sirolimus is coated on the surface of the stent and the balloon (108µg of sirolimus on a 3.0×16.0mm system). Sirolimus is programmed to be completely released within 28 days, however, the tissue concentration of sirolimus peakes within the first 24h. A preclinical study with the FOCUS np stent showed similar LLL and inflammation scores to that seen in the Cypher SES at 28 days and at 90 days. A FIM trial will be completed in early 2013.38
 images of the crimped stent (A) and the magnified surface of the strut and balloon (B) coated with encapsulated sirolimus (C, yellow arrows). The nanocarrier consists of a lipid bilayer with a hydrophilic head and two lipophilic/hydrophobic tails (D), and the drug is released on pH change.")
The nanocarrier-based FOCUS np sirolimus-eluting stent. The scanning electron microscopy (SEM) images of the crimped stent (A) and the magnified surface of the strut and balloon (B) coated with encapsulated sirolimus (C, yellow arrows). The nanocarrier consists of a lipid bilayer with a hydrophilic head and two lipophilic/hydrophobic tails (D), and the drug is released on pH change.
Fully bioresorbable scaffolds (BRS) are a novel approach as they provide transient vessel support in contrast to the permanent caging caused by metallic stents. The concept of BRS was introduced by Stack et al. in the year 1988.39 Zidar and colleagues first implanted BRS made of poly-L-lactic acid (PLLA) into canine femoral arteries.40 Despite significant scaffold degradation with low-grade vascular inflammation at 9-month follow-up, this technology failed to develop because of the inability to manufacture an ideal polymer that could limit inflammation and restenosis.41,42 In the year 2000, Tamai and colleagues reported their FIM experience with BRS implantation for the treatment of human coronary arteries.43 This “Igaki-Tamai” PLLA stent had a unique zigzag helical coil design, with a strut thickness of 170µm. This system was self-expanding but also required balloon inflation with heated contrast for expansion. The FIM study of the Igaki-Tamai stent (15 patients) demonstrated no MACE or ST events within 30 days, and 1 repeat percutaneous coronary intervention at 6-month follow-up. Our group reported the findings of OCT at 10 years after Igaki-Tamai stent implantation, showing absence of visible struts, with endoluminal lining of the vessel wall.44 Recently, Nishio et al. reported >10-year clinical outcomes of the first 50 patients treated with Igaki-Tamai stents. Autopsy specimens showed interesting histological findings, that indicated healing with thickened neointima at the previously stented segment, without inflammatory cell infiltration or foreign body reactions. As measured by quantitative coronary angiography, LLL decreased from 0.91 mm at 6 months to 0.59mm at 3 years, whilst intravascular ultrasound (IVUS) showed an increased external elastic lamina area (15.0mm2 postprocedurally and 16.9mm2 at 3 years). These findings suggest that the artery restored its capability to respond to expansive remodeling and late lumen enlargement once the scaffold degraded.
Currently, numerous BRS are being tested in clinical or preclinical studies. An overview of this technology has been shown in Table 3 and Figure 5.
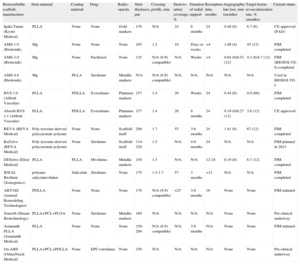
Overview of Bioresorbable Scaffolds Under Current Pre-clinical or Clinical Investigation
| Bioresorbable scaffold, manufacturer | Strut material | Coating material | Drug | Radio- opacity | Strut thickness, µm | Crossing profile, mm | Stent-to-artery coverage, % | Duration of radial support | Resorption time, months | Angiographic late loss, mm (months) | Target lesión revascularization rate, % (months) | Current status |
| Igaki-Tamai (Kyoto Medical) | PLLA | None | None | Gold markers | 170 | N/A | 24 | 6 months | 24 | 0.48 (6) | 6.7 (6) | CE approved (PAD) |
| AMS-1.0 (Biotronik) | Mg | None | None | None | 165 | 1.2 | 10 | Days or weeks | <4 | 1.08 (4) | 45 (12) | FIM completed |
| AMS-3.0 (Biotronik) | Mg | None | Paclitaxel | None | 125 | N/A (6 Fr compatible) | N/A | Weeks | >4 | 0.64 (6)0.52 (12) | 4.3 (6)4.7 (12) | FIM (BIOSOLVE-I) completed |
| AMS-4.0 (Biotronik) | Mg | PLLA | Sirolimus | Metallic markers | N/A | N/A (6 Fr compatible) | N/A | N/A | N/A | N/A | N/A | Used in BIOSOLVE-I |
| BVS 1.0 (Abbott Vascular) | PLLA | PDLLA | Everolimus | Platinum markers | 157 | 1.4 | 26 | Weeks | 24 | 0.44 (6) | 0.0 (60) | FIM completed |
| Absorb BVS 1.1 (Abbott Vascular) | PLLA | PDLLA | Everolimus | Platinum markers | 157 | 1.4 | 26 | 6 months | 24 | 0.19 (6)0.27 (12) | 3.6 (12) | CE approved |
| REVA (REVA Medical) | Poly-tyrosine-derived polycaronate polymer | None | None | Scaffold itself | 200 | 1.7 | 55 | 3-6 months | 24 | 1.81 (6) | 67 (12) | FIM completed |
| ReZolve (REVA Medical) | Poly-tyrosine-derived polycaronate polymer | None | Sirolimus | Scaffold itself | 114-228 | 1.5 | N/A | 4-6 months | 24 | N/A | N/A | FIM planned in 2013 |
| DESolve (Elixir Medical) | PLLA | PLLA | Mvolimus | Metallic markers | 150 | 1.5 | N/A | N/A | 12-24 | 0.19 (8) | 6.7 (12) | FIM completed |
| IDEAL BioStent (Xenogenics) | polymer salicylate+linker | Salicylate | Sirolimus | None | 175 | 1.5-1.7 | 57 | 3 months | >12 | N/A | N/A | FIM completed |
| ART18Z (Arterial Remodeling Technologies) | PDLLA | None | None | None | 170 | N/A (6 Fr compatible) | <25 | 3-6 months | 18 | None | None | FIM initiated |
| Xinsorb (Huaan Biotechnology) | PLLA+PCL+PLGA | None | Sirolimus | Metallic markers | 160 | N/A | N/A | N/A | N/A | None | None | Pre-clinical underway |
| Amaranth PLLA (Amaranth Medical) | PLLA | None | None | None | 150-200 | N/A (6 Fr compatible) | N/A | 3-6 months | N/A | None | None | FIM initiated |
| On-ABS (OrbusNeich Medical) | PLLA+PCL+PDLLA | None | EPC+sirolimus | None | 150 | N/A | N/A | N/A | N/A | None | None | Pre-clinical underway |
CE, Conformité Européenne; EPC, endothelial progenitor cell (capture technology); FIM, first-in-man; Mg, magnesium; N/A, not applicable; PAD, peripheral artery disease; PCL, poly-L-lactide-co-e-caprolactone; PDLLA, poly-D, L-lactic acid; PLGA, poly-lactide-co-glycolide; PLLA, poly-L-lactic acid.

Magnesium (Mg) is the fourth commonest cation within the human body. It is essential for the synthesis of over 300 enzymes, and is a cofactor for ATPase. A high dose infusion of Mg can cause vasodilatation and the development of collaterals during ischaemia. The degradation of Mg produces an electronegative charge that results in the stent being hypothrombogenic.45
The first generation absorbable metallic stent (AMS-1, Biotronik; Berlin, Germany) was composed of 93% Mg and 7% rare earth metals. In the porcine model, the AMS-1 was shown to be rapidly endothelialized, and largely degraded into inorganic salts at 60 days, with little associated inflammatory response.46 The PROGRESS AMS trial was a signle-arm FIM study, which assessed the efficacy and safety of this stent in 63 patients with single de novo lesions.47 No evidence of death, MI, or ST was reported at up to 12-month follow-up. Disappointingly, the TLR rate was 23.8% at 4 months and 45% at 12 months. The in-stent LLL was 1.08mm and the vasodilator function, after the nitroglycerin administration, appeared to be restored in the stented segment at 4-month angiographic follow-up.48 IVUS data suggested that the increased LLL was attributed to an increased neointimal formation and insufficient radial strength of the Mg alloy, due to rapid stent degradation resulting in vessel recoil. Consequently, new stents have been developed, namely AMS-2 and AMS-3. The AMS-2 stent was designed to address excessive vessel recoil seen with AMS-1. It provided prolonged mechanical integrity by using a different Mg alloy, which not only had a higher collapse pressure, but also a slower degradation time. In addition, the strut thickness was reduced from 165µm to 125µm, and the cross-sectional shape of the strut altered from rectangular to square. These modifications facilitated prolonged mechanical integrity, improved radial strength, and resulted in reduced neointimal proliferation in animal studies. The AMS-3 stent (ie, drug-eluting AMS [DREAMS]) was designed to incorporate a bioresorbable matrix for the controlled release of paclitaxel with the AMS-2 platform. This device was evaluated in the BIOSOLVE-I trial (46 patients), and demonstrated an in-stent LLL of 0.64mm at 6 months and 0.52mm at 12 months. The rate of TLF was 7.0% at up to 12-month follow-up, due to 2 clinically driven TLRs and 1 periprocedural MI.49 The second generation DREAMS has a modified stent platform and sirolimus as its antiproliferative drug. The BIOSOLVE-II study aimed to assess the safety and efficacy of this device will be initiated in the year 2013.
Everolimus-eluting Poly-L-lactic Acid Scaffold: Absorb BVSThe backbone of Absorb BVS (Abbott Vascular; Santa Clara, California, United States) is made of PLLA. The coating consists of poly-D, L-lactide (PDLLA), which is a random copolymer of D-lactic acid and L-lactic acid with lower crystallinity than the backbone PLLA. The PDLLA coating controls the release of the antiproliferative drug everolimus. The first generation Absorb BVS (1.0) was tested in 30 patients who were enrolled in the ABSORB FIM (cohort A) trial. Multiple modality imaging was assessed in this trial, and the results can be summarized as follows: a) partial bioresorption of the polymeric struts; b) late lumen enlargement between 6 months and 2 years; c) restoration of vasomotion and endothelial function at 2 years; d) sustained scaffolding of plaque deformability documented with palpography, and e) feasibility of noninvasive imaging with multislice computed tomography.50,51 Five-year clinical follow-up is available in 29 patients.52 Only 1 patient experienced a non-Q wave MI related to the treatment of a non-flow-limiting stenosis at 46 days after Absorb BVS implantation. There were no ST events in the entire period and no MACE between 6 months and 5 years, resulting in an overall MACE rate of 3.4% at 5 years. Late scaffold shrinkage was the primary reason for an increased in-stent LLL (0.44mm) at 6 months. Lumen area was reduced by 16.6%, whilst the late recoil was 11.7%.53 In order to enhance the radial strength of the struts and to reduce late recoil, the strut design and the manufacturing process of the polymer were modified in the second generation Absorb BVS (1.1). Firstly, the new design had in-phase zigzag hoops linked by bridges that allowed for a more uniform strut distribution. This new scaffold design reduced maximum circular unsupported surface area that provided for more uniform vessel wall support and drug delivery. Secondly, a modified manufacturing process resulted in a slower hydrolysis (in vivo degradation) rate of the polymer, thus allowing for prolongation of its mechanical integrity.54
The Absorb BVS 1.1 was evaluated in 101 patients in the ABSORB cohort B trial. This cohort was divided in 2 subgroups: the first group (B1) underwent invasive imaging with quantitative coronary angiography, IVUS, and OCT postprocedurally, at 6 months, and at 24 months; whereas the second group (B2) underwent invasive imaging postprocedurally, at 12 months, and at 36 months. In the entire cohort B population, the overall MACE rate was 9.0%, including 3 non-Q wave MIs and 6 ischemia-driven TLRs, without cardiac death during the 2-year follow-up. There were no possible, probable, or definite scaffold thromboses despite dual antiplatelet therapy rates of 97% at 6 months, 81.2% at 12 months, and 24.8% at 24 months.
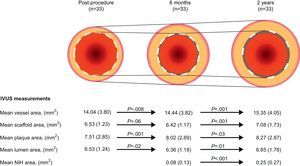
For the cohort B1 population, serial multimodality imaging results are currently available.55 Serial angiographic analyses showed that in-scaffold LLL of 0.16mm at 6 months increased to 0.27mm at 2 years. Notably, serial IVUS analyses demonstrated that the mean lumen area increased, whereas the minimum lumen area remained stable between 6 months and 2 years (Fig. 6). Percentage hyperechogenic area, a more sensitive parameter to measure degradation of polymeric material, decreased from 25.3% postprocedurally to 20.4% at 6 months and to 13.8% at 2 years. Similar to IVUS, serial OCT investigation confirmed the progressive increase in mean scaffold area from 7.47mm2 postprocedurally, to 7.70mm2 at 6 months, and 8.24mm2 at 24 months.

The promising results of Absorb BVS constitute the proof of concept that this device can adequately revascularize coronary vessels and prevent restenosis. The Absorb BVS acquired the CE mark approval in January 2011, and since September 2012 it is commercially available in different diameters (2.5mm, 3.0mm, and 3.5mm) and lengths (12mm, 18mm, and 28mm). This device is now being evaluated in the ABSORB-EXTEND registry (~800 patients). A pivotal, randomized trial (ABSORB II), comparing Absorb BVS with Xience Prime EES (Abbott Vascular) in 500 patients, is simultaneously ongoing in Europe.
Tyrosine Polycarbonate StentThe REVA stent (REVA Medical, San Diego, California, United States) consists of a tyrosine-derived poly carbonate that degrades into water, carbon dioxide, and ethanol. In addition to its radio-opacity, the REVA stent also has a unique “slide and lock” design that provides flexibility. This design maintains the acute lumen gain following stent deployment, and provides additional support to the scaffold during vessel remodeling. The RESORB FIM trial enrolled 27 patients. The in-stent LLL was disappointingly high (1.81mm) and IVUS data showed no vessel recoil as indicated by external elastic lamina area (15.5mm2 postprocedurally and 15.3mm2 at 6 months). There was a high rate of TLR (66.7%) between 4 months and 6 months, mostly due to excessive neointimal hyperplasia.56 The second generation ReZolve stent had a more robust polymer, a “spiral” slide and lock system, and a coating of sirolimus. Furthermore, the ReZolve2 stent had a smaller profile (1.52mm) and achieved approximately 30% increase in radial strength. The safety and efficacy of the ReZolve or ReZolve2 stent is currently under investigation in the RESTORE study (50 patients) that was initiated in December 2011. Preliminary data (26 patients) showed that technical success rate was 85%, due to the delivery failure seen in 4 patients. Two cases with TLR as a primary endpoint were reported at 6-months follow-up.57
Myolimus-eluting Poly-L-Lactic Acid Scaffold: DESolveDESolve BRS (Elixir Medical; Sunnyvale, California, United States) has a similar PLLA backbone to the Absorb BVS, but it is coated with myolimus (3µg/mm), an mTOR inhibitor macrocyclic lactone, and a sirolimus analogue. Sufficient radial strength was achieved over 3 months and the bioresorption of the scaffold was observed between 1 year and 2 years. The DESolve-I FIM trial (16 patients) demonstrated that the rate of acute recoil was 6.4%, and in-scaffold LLL was 0.19 mm at 6 months.58 In IVUS investigation, the respective mean scaffold area and lumen area was 5.35mm2 and 5.35mm2 postprocedurally, and 5.61mm2 and 5.10mm2 at 6 months (P=no significant). OCT revealed that 98.7% of the struts were covered at 6 months. All patients were clinically followed up to 1 year, and 3 patients experienced MACE including 1 cardiac death, 1 target vessel MI, and 1 TLR. There were no patients with the evidence of ST. The DESolve Nx trial is currently enrolling 120 patients treated with the next generation DESolve Nx stent with novolimus (5µg/mm), which is an active metabolite of sirolimus.59
Poly Salicylic Acid Stent: IDEALThe IDEAL BRS (Xenogenics Corp.; Canton, Massachusetts, United States) has 2 components: the backbone and the drug layer. The backbone of the device is made of polylactide anhydride mixed with a polymer of salicylic acid and sebacic acid linker. The drug layer consists of salicylate that controls the release of the antiproliferative drug sirolimus. The presence of salicylic acid provides the device with anti-inflammatory properties, which have been confirmed in preclinical studies.60 The IDEAL BRS was tested in a small number of humans (11 patients) in 2008. Although this study has not been fully reported, there was insufficient neointimal suppression and a reduction in lumen area due to inadequate drug dosing and rapid release of the sirolimus.61 The second generation IDEAL BioStent has a higher drug dose, slower drug-release kinetics, and a smaller system profile. The device is currently undergoing preclinical evaluation.
Arterial Remodeling Technologies Bioresorbable ScaffoldThe ART BRS (Arterial Remodeling Technologies; Noisy le Roi, France) is manufactured from a PDLLA amorphous polymer without an antiproliferative drug. This device is 6 Fr compatible, and provides transient scaffolding for 5 months to 7 months. Full resorption occurs within 18 months. The performance of the ART BRS was compared with the BMS in rabbit and porcine models, and no MACE was reported. The acute recoil was comparable to BMS. Interestingly, angiographic analyses demonstrated the phenomenon of late lumen enlargement, as well as increased external elastic lamina area detected by IVUS at 9 months. Based on the results of preclinical studies, the ARTDIVA FIM trial has already commenced and is recruiting patients at 5 sites in France. It aims to evaluate clinical outcomes at 6 months.62
Xinsorb Bioresorbable ScaffoldThe Xinsorb BRS (Huaan Biotechnology; Laiwu, China) is a fully bioresorbable sirolimus-loaded scaffold that consists of PLLA, poly-lactide-co-glycolide, and poly-L-lactide-co-e-caprolactone. An experimental study evaluated the feasibility of Xinsorb BRS in comparison with the Excel DES (JW Medical; Shandong, China). Sixteen Xinsorb scaffolds and 16 Excel stents were implanted in the coronary arteries of porcine models.63 In vitro drug-elution kinetics indicated that 78% of sirolimus was released from Xinsorb BRS within the first 14 days. Histomorphometry demonstrated a significantly lower percentage diameter restenosis in the Xinsorb BRS compared to the Excel DES (18.6% vs 21.4% at 30 days and 24.5% vs 27.7% at 90 days, respectively). In addition, the struts of the Xinsorb BRS were completely covered by neointima at 90 days.64 Although these preliminary results are encouraging, further extensive preclinical studies are necessary to investigate the safety and efficacy of this device. The company is expecting to organize a FIM trial in the year 2013.
Other Brioresorbable ScaffoldsThe Amaranth PLLA scaffold (Amaranth Medical; Mountain View, California, United States) and the On-ABS (OrbusNeich, Hong Kong, China) are currently under preclinical evaluation. In addition, there are several other devices that are still under development. These include the Sahajanand BRS (Sahajanand Medical Technologies; Surat, India), the Avatar BRS (S3V; Hyderabad, India), the MeRes BRS (Meril Life Sciences; Vapi, Gujarat, India), and the Zorion BRS (Zorion Medical; Indianapolis, Indiana, United States).
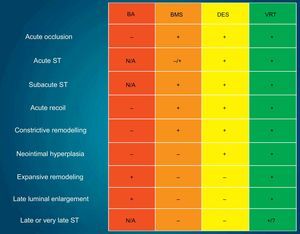
FUTURE PERSPECTIVESThe new enemy in the DES era—ST—has accelerated technological evolution in interventional cardiology. Newer generation DES, with biodegradable polymers, have shown an impressive reduction in VLST, contributing to improved long-term outcomes, as compared to first generation DES. The polymer-free DES or BRS are relatively new technologies, with many trials still in progress. The currently available angiographic, intravascular imaging, and clinical data, suggest acceptable safety and efficacy of these new technologies. However, it remains unclear as to whether biodegradable-polymer DES or polymer-free DES can minimize late ST events, particularly as these late events have also been observed in patients receiving BMS.65,66 Furthermore, considering the fatal consequences of ST, focus should be maintained on the eradication, rather than the minimization of this serious complication. There is a fundamental difference in concept between the DES and the BRS technologies, with the latter having a capability of liberating the vessel from a permanent metallic cage. Therefore, BRS technology has a theoretical advantage in reducing ST by means of endoluminal prosthesis elimination. BRS also facilitates the restoration of vasomotor function, which indirectly results in the completeness of vessel healing. The entire process of this treatment has been hence named as vascular reparative therapy (Fig. 7).67

Schematic illustration presenting the evolution of percutaneous coronary interventions. BA, balloon angioplasty; BMS, bare metal stents; DES, drug-eluting metallic stents; N/A, not applicable because of the absence of stent; ST, stent thrombosis; VRT, vascular reparative therapy. “+” implies prevented or not restricted, whilst “-” implies not prevented or restricted. Modified with permission from Serruys et al.67
One possible fate of the stenotic lesion treated with metallic stents is the development of in-stent neointimal tissue (even seen with DES), where the antiproliferative drug slows down or postpones the phenomenon. This neointimal tissue may in turn become atherosclerotic, degenerate to a vulnerable plaque, and finally rupture inside the cage of the stent (ie, neoatherosclerosis).68,69 A stiff metallic stent can also alter vessel geometry and biomechanics, which may result in long-term flow disturbances and chronic irritation, in addition to the risk of late strut fractures, which potentially contribute to restenosis and adverse clinical events.70,71 From a physiological perspective, the absence of a rigid metallic cage facilitates the restoration of vasomotor function, adaptive shear stress, and late luminal enlargement. After bioresorption, there would be no triggers for thrombosis, such as uncovered struts or durable polymers. The absence of foreign materials may also reduce concerns about future treatment options, such as precluding bypass-graft surgery, and the requirements for long-term dual antiplatelet therapy with a potential reduction in associated bleeding complications. Since BRS have only been evaluated in limited patients with noncomplex lesions, the feasibility of these devices in complex lesions requires further clinical evaluation. In addition, future investigations are required to establish if BRS technology is superior to permanent metallic DES.
CONCLUSIONSNewer metallic DES technology has proven to decrease the risk of revascularization and ST events. The optimal design, however, of scaffolds, polymers, antiproliferative drugs and their degradation/release kinetics is still under investigation. BRS technology is anticipated not only to eliminate the risk of VLST, but also to contribute to the restoration of physiological function of treated vessels. Although further technical development and clinical evaluation are required before BRS can be accepted as the ultimate device for the treatment of coronary artery disease, this new technology looks promising and could be the next revolution in interventional cardiology.
CONFLICTS OF INTERESTNone declared.
.