
There are few reports of the appearance of pulmonary arterial hypertension following arterial switch surgery in the neonatal period to correct transposition of the great arteries. We assessed the frequency and clinical pattern of this complication in our series of patients.
MethodsOur database was reviewed to select patients with transposition of the great arteries corrected by neonatal arterial switch at our hospital and who developed pulmonary hypertension over time.
ResultsWe identified 2 (1.3%) patients with transposition of the great arteries successfully repaired in the first week of life who later experienced pulmonary arterial hypertension. The first patient was a 7-year-old girl diagnosed with severe pulmonary hypertension at age 8 months who did not respond to medical treatment and required lung transplantation. The anatomic pathology findings were consistent with severe pulmonary arterial hypertension. The second patient was a 24-month-old boy diagnosed with severe pulmonary hypertension at age 13 months who did not respond to medical therapy.
ConclusionsPulmonary hypertension is a rare but very severe complication that should be investigated in all patients with transposition of the great arteries who have undergone neonatal arterial switch, in order to start early aggressive therapy for affected patients, given the poor therapeutic response and poor prognosis involved.
Keywords
The onset of pulmonary arterial hypertension (PAH) in patients with d-transposition of the great arteries (d-TGA) is a potentially severe complication first described decades ago.1–3 It usually affects patients with d-TGA not corrected surgically,1 particularly those with associated ventricular septal defect and/or ductus arteriosus, in whom continuous exposure to increased pulmonary flow eventually causes vascular remodeling and the resulting increase in pulmonary vascular resistances.4 Following the introduction of atrial switch surgery, the incidence of this complication was greatly reduced,5 and once the arterial switch (AS) operation was established as the surgical repair method of choice in the 1980s, there have been few reports of PAH in patients with d-TGA.6–9 The pathophysiologic mechanism is unknown in this type of PAH, which recurs or appears months or years after AS in the absence of significant residual lesions, and it has an aggressive course.6–9 Some studies report it as the most common cause of late mortality after this type of surgical procedure.10
The aim of this study was to evaluate the incidence of PAH after neonatal surgical repair of d-TGA by AS in our series of patients and to describe the clinical and hemodynamic characteristics of 2 cases of PAH after AS surgery.
METHODSA retrospective study was performed at a third-level referral hospital for cardiac surgery in congenital heart disease between January 1995 and December 2014. Using our database, information was collected on all patients with simple or complex forms of d-TGA who had undergone AS during the neonatal period, considered the first 6 weeks of life. Patients with complex d-TGA who had undergone a surgical procedure other than AS were excluded. Following surgical repair, all patients underwent follow-up in the outpatient clinic at our hospital, including medical history, physical examination, electrocardiogram, and transthoracic echocardiogram. When pulmonary hypertension was suspected, cardiac catheterization was also performed. All patients were diagnosed and treated in accordance with current clinical guidelines.11,12
RESULTSOur study population was composed of 152 patients with d-TGA corrected neonatally by AS: 96 (63.2%) patients with d-TGA who had an intact interventricular septum and 56 (36.8%) who had complex forms of d-TGA (in combination with associated ventricular septal defect and/or aortic coarctation). After a mean of 8.5 ± 4.7 years (range, 10 months-19 years) after surgery, 2 (1.3%) patients experienced PAH. The characteristics of the patients affected and their clinical course are listed in Table 1.
Characteristics, Clinical Course, and Outcomes of Patients Who Develop Pulmonary Arterial Hypertension After Neonatal Arterial Switch Surgery
| Patient | A | B |
|---|---|---|
| Type of TGA | TGA with intact septum | TGA + VSD |
| Age at AS, days | 6 | 7 |
| Surgical procedure | AS | AS + VSD closure |
| Age at PAH diagnosis, months | 7 | 13 |
| Cardiac catheterization | PAP, 74/7 (39) mmHg mPAP/mAP, 0.96 PVR, 14 UW/m2 | PAP, 85/6 (41) mmHg mPAP/mAP, 0.78 PVR, 15 UW/m2 |
| Treatment | Bosentan + sildenafil + inhaled iloprost | Bosentan + sildenafil |
| Clinical course | Lung transplantation at age 3 y + 10 months | Clinically stable at age 24 mo; NYHA functional class II |
AS, arterial switch; mAP, mean aortic pressure; mPAP, mean pulmonary artery pressure; NYHA, New York Heart Association; PAH, pulmonary arterial hypertension; PAP, pulmonary artery pressure; PVR, pulmonary vascular resistance; TGA, transposition of the great arteries; VSD, ventricular septal defect.
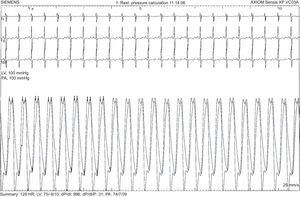
The first patient was a 7-year-old girl with a prenatal diagnosis of d-TGA with intact interventricular septum who underwent AS surgery at age 6 days with no postoperative complications or residual lesions. After an initial favorable clinical course, at 7 months of age she showed failure to thrive and 90% oxygen saturation. Echocardiography showed a small interatrial defect with 2-way shunt and signs of pulmonary hypertension. Cardiac catheterization confirmed systemic pulmonary pressure with high pulmonary vascular resistances and a positive pulmonary vasodilator test (Figure 1). No other abnormalities were found to explain the clinical picture. Because the patient was younger than 1 year (a relative contraindication for calcium blockers) and had an aggressive hemodynamic profile of pulmonary hypertension, combination therapy with sildenafil and bosentan was initiated. After 6 months of treatment, inhaled iloprost was added, but the patient's situation progressively worsened, and at age 3 years and 10 months, she underwent lung transplantation. Anatomic pathology of the native lung showed findings consistent with PAH and irreversible tissue damage (Figure 2). At 4 years posttransplant, the patient was clinically stable.

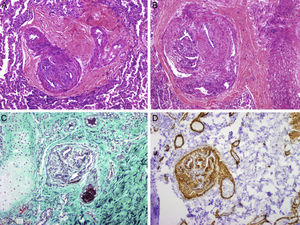
. C, concentric fibrosis of the wall was also observed in the right arteriole, as well as images of plexiform lesions with proliferation of endothelial, smooth muscle, and myofibroblast cells that form intramural secondary lumens, particularly in the areas of vessel bifurcation (B, hematoxylin-eosin; D, smooth muscle actin) (courtesy of Dr. J.C. Ferreres, Hospital Vall d’Hebron, Barcelona).")
Pulmonary histopathology. Images showing obstructive pulmonary vascular disease: intimal hyperplasia in a muscular artery with intimal collagenation and lumen diameter reduction (A, hematoxylin-eosin; C, Masson trichrome). C, concentric fibrosis of the wall was also observed in the right arteriole, as well as images of plexiform lesions with proliferation of endothelial, smooth muscle, and myofibroblast cells that form intramural secondary lumens, particularly in the areas of vessel bifurcation (B, hematoxylin-eosin; D, smooth muscle actin) (courtesy of Dr. J.C. Ferreres, Hospital Vall d’Hebron, Barcelona).
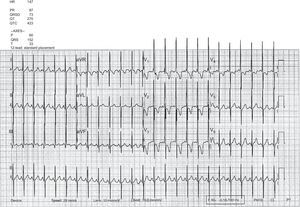
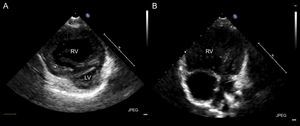
The second patient was a 2-year-old boy with a prenatal diagnosis of d-TGA and ventricular septal defect who underwent AS and surgical closure of a ventricular septal defect at age 7 days, with no postoperative complications or residual lesions. He remained asymptomatic until age 13 months, when he began to experience progressive dyspnea. The electrocardiogram showed signs of significant enlargement of the right chambers with a strain pattern (Figure 3). Transthoracic echocardiography disclosed severe pulmonary hypertension, with considerable right ventricular dilation and left ventricular compression (Figure 4). Cardiac catheterization revealed elevated pulmonary pressure and pulmonary vascular resistances, with a negative pulmonary vasodilator test. Once sildenafil and bosentan therapy was started, the patient's clinical situation improved. At the time of writing, he remained in functional class II, although echocardiographic signs of severe PAH persisted.


The earliest cases of PAH associated with d-TGA were reported by several autopsy series of patients who had not undergone surgery. In these series, PAH was more frequent in older patients and in those with higher pulmonary flows.1–4 These findings are similar to those of a pooled analysis of various series,4 which showed an increasing rate of Heath and Edwards grade ≥ 3 hypertensive pulmonary vascular disease13 with age, reaching 34% among patients aged > 12 months. There are also reports of persistent severe pulmonary hypertension in newborns with TGA,14,15 which indicates that pulmonary vascular disease progression is early and rapid in this heart condition.
Following the introduction of atrial switch surgery, which was performed at a mean age of 5 to 22.1 months, the incidence of this complication dropped to 3.5% to 19%,5,6,16,17 and once neonatal AS became the corrective surgery of choice, reports of the subsequent appearance of PAH became rarer,6–9 indicating a greater possibility of hypertensive pulmonary vascular disease with longer exposure to the circulatory abnormality of the transposition of the great arteries. After AS, PAH has a frequency of around 1%,9,18 similar to our result of 1.3%, and even affects patients without long preoperative exposure to increased pulmonary flow (ie, without long postnatal exposure to the transposition physiology) and with no postoperative residual lesions,6,8,9 as in the 2 patients in our series who underwent early surgery. These findings indicate that PAH progression is more rapid in d-TGA than in other congenital heart diseases and that pulmonary vascular disease sometimes appears very early, possibly during the fetal period. Various hypotheses have been proposed to explain accelerated PAH development in patients with d-TGA. The most widely accepted theory suggests that pulmonary vasoconstriction secondary to hypoxemia, increased pulmonary flow, and the arrival of poorly oxygenated blood from bronchopulmonary collateral circulation would be the main triggers.19 However, this would not explain PAH in patients who have undergone neonatal AS, whose main trigger may be flow pattern abnormalities in fetal circulation. The arrival of oxygen-rich blood to the pulmonary artery and to the ductus arteriosus from the umbilical vein through the inferior vena cava causes ductal constriction and, therefore, the arrival of a greater blood volume at a higher pressure to pulmonary circulation.16 This prenatal factor is also listed in the pediatric pulmonary hypertension classification from the Panama group,20 which includes PAH among patients with d-TGA among prenatal or developmental pulmonary vascular disease. At our hospital, prenatal studies are not routinely performed on the restrictive nature of foramen ovale and ductus arteriosus constriction because the various echocardiographic parameters proposed to date do not have good predictive power.21,22
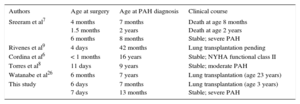
Although the pathophysiologic mechanism causing the pulmonary vascular abnormality is unknown, it is evident that patients with d-TGA are prone to PAH, probably in relation to a permissive genotype,23 and the risk is not reduced after surgical correction. For this reason, it is difficult to classify this type of PAH which, according to Niza's classification,24 could be included among postoperative PAH or PAH consistent with congenital heart disease. This classification includes patients with PAH of similar behavior to that of idiopathic PAH, poor response to medical therapy, and a poor prognosis, with an incidence of 1 to 2 cases per million inhabitants per year among the general population,25 a common outcome reported among d-TGA patients published to date6–9,26 (Table 2).
Patients With Pulmonary Arterial Hypertension After Correction of Transposition of the Great Arteries by Arterial Switch
| Authors | Age at surgery | Age at PAH diagnosis | Clinical course |
|---|---|---|---|
| Sreeram et al7 | 4 months 1.5 months 6 months | 7 months 2 years 8 months | Death at age 8 months Death at age 2 years Stable; severe PAH |
| Rivenes et al9 | 4 days | 42 months | Lung transplantation pending |
| Cordina et al6 | < 1 months | 16 years | Stable; NYHA functional class II |
| Torres et al8 | 11 days | 9 years | Stable; moderate PAH |
| Watanabe et al26 | 6 months | 7 years | Lung transplantation (age 23 years) |
| This study | 6 days 7 days | 7 months 13 months | Lung transplantation (age 3 years) Stable; severe PAH |
NYHA, New York Heart Association; PAH, pulmonary arterial hypertension.
The postoperative period until the diagnosis of PAH varied from 7 months to 16 years.6,7 In our series, the 2 affected patients are among the youngest patients reported to date (PAH diagnosis at 7 and 13 months, respectively) and among those with an aggressive clinical course: the first had a poor response to medical therapy and lung transplantation at age 3 years, with Heath and Edwards grade 4 lesions on anatomic pathology, and the second showed echocardiographic evidence of severe PAH despite pulmonary vasodilator therapy.
CONCLUSIONSThe appearance of PAH after surgical repair consisting of AS in patients with d-TGA is a rare but potentially very severe complication that has characteristics similar to those of idiopathic PAH and greatly impacts the morbidity and mortality of affected patients. Close follow-up is essential in all patients who have undergone neonatal AS to identify any PAH and to establish early aggressive therapeutic measures, in view of the strong probability of a rapid and aggressive clinical course.
CONFLICTS OF INTERESTNone declared.
- –
The appearance of PAH associated with transposition of the great arteries is a severe complication which has become rarer with the introduction of new and earlier surgical techniques, such as AS. However, the etiology and incidence of the appearance of PAH after this type of surgical repair are unknown.
- –
The mid-term incidence of PAH after AS surgery is 1.3%. The hemodynamic and clinical profile is similar to that of idiopathic PAH, with an aggressive clinical pattern and severe prognosis; hence, early diagnosis and early intensive therapy are essential.