
Keywords
INTRODUCTION
Pulmonary hypertension complicates the courseof many children and adults with congenital heartdiseases (CHD). The increase in pulmonary pressureassociated with CHD is either secondary to increasedpulmonary blood flow or to increased post-capillarypressures. Pulmonary arterial hypertension (PAH) is in the vast majority associated to congenitalshunts.
Despite major advances in the understanding ofthe regulation of the pulmonary vascular bed and thepulmonary endothelial lesions leading to pulmonaryvascular disease, despite the advances in surgicalrepair and the discovery of potential therapiesin the pre and postoperative period, pulmonaryhypertension still carries a significant mortality andmorbidity in patients with CHD.
One of the most important aspects that needsto be defined is the exact wording used to definethe disease in the setting of PAH associated withCHD. Based on hemodynamic definition of PAH(mean pulmonary arterial pressure [PAP] >25mmHg)1 almost all patients present with pulmonaryhypertension in the presence of a large unrestrictedleft to right shunt, but what is important in thissetting is the degree of pulmonary vascular lesionsand what would be called pulmonary vasculardisease (PVD). Indeed, a patient with highpulmonary blood flow and low pulmonary vascularresistance (PVR) will fulfil the requirements for adiagnosis of PAH but can be definitely treated withsurgical closure of the shunt. On the contrary, apatient with low pulmonary blood flow, cyanosiswith reverted shunt (right to left) and high PVR,and the so called Eisenmenger's syndrome (ES) willnot benefit from surgical closure and this is evencontraindicated, but will potentially benefit fromnew targeted therapies for PAH.
The recent introduction of targeted therapies inother forms of PAH has led to a renewed interest inpulmonary hypertension associated with CHD andthis particularly for the most advanced form, theES.2
The particular setting of the single ventriclephysiology is also of major interest as even a minimalincrease in pulmonary vascular resistance may eitherpreclude Fontan surgery or lead to failure of thiscirculation.3
This review summarizes the current knowledgeon pulmonary hypertension associated withCHD characterized by congenital shunts. Specificdiscussions will be dedicated to pre and postoperativePAH, ES management and the particular setting ofthe Fontan circulation.EPIDEMIOLOGY AND CLASSIFICATION
A wide range of CHD can lead to PAH butthe most important group is left-to-right shuntslesions. This group includes many differentdefects that have different evolutions and this isof importance.
Advances in pediatric cardiology and surgeryhave increased the number of CHD patientssurviving into adulthood, and helped to preventthe onset of ES in many patients in the westernworld, resulting in a reduction of approximately50% in prevalence over the past 50 years. Around5% of adults with CHD will develop PAH.4 Theprevalence of PAH in CHD has been estimatedbetween 1.6 and 12.5 million of adults, with 25-50% presenting with ES.5 However, still a growingnumber of patients present with malformationscharacterized by a so-called single ventriclephysiology requiring a particular surgical approach(partial or total cavopulmonary anastomosis).Although they do not present pulmonaryhypertension as per the classical definition, thesepatients may have pulmonary vascular lesionsthat either preclude surgery or carry a high risk ofmorbidity and mortality.
Structural changes in the pulmonaryvasculature in all forms of PAH, including ES,are qualitatively similar, although there is somevariation in the distribution and prevalence ofpathological changes with different underlyingetiologies. According to the classification,pulmonary hypertension resulting from CHD isgrouped with idiopathic/heritable PAH, drug-related PAH, PAH associated with connectivetissue diseases and HIV-related PAH.6 However,CHD is, as mentioned above, a complex groupof pathologies that may differ from other formsof PAH with regards to cardiac anatomy,hemodynamic and natural history.7, 8 This is one of the reasons why experts have tried progressivelyto develop a sub-classification to better definePAH-CHD patients.8-10 Sub-classifications havetaken into account several factors importantto better describe the lesions but also factorsimportant in the development of PVD such astype and size of defects, hemodynamic, presenceof extra cardiac anomalies and the status of therepair (unrepaired, palliated or repaired). Basedon these suggestions and further understanding of the disease, several modifications have beenintroduced at the last world meeting on pulmonaryhypertension allowing for an updated pathologicand physiopathology classification that shouldsatisfy both the CHD expert and non expert.6
For clinical practice use, four distinctphenotypes have been recognised, differing intheir management and responses to treatment(Table 1). The first group is composed of patientswith ES, at the final stage of PAH-CHD butbeneficiating from the emerging therapies. Thesecond cluster encompasses patients with PAHassociated with systemic-to-pulmonary shunts,at earlier stages of the disease. These pre-Eisenmenger patients have a mildly or moderatelyincreased PVR. Unlike ES patients, they are notoften enlisted in studies and their management isconsequently challenging.

The third group includes patients with a smallcardiac shunt that is not thought to be the causeof PAH. They are very similar to idiopathic PAHpatients.
The last group is composed of patients with apersistent or recurrent PAH after successful surgicalcorrection of the cardiac defect. Their very poorprognosis emphasizes the need for more accurateoperability criteria.
These different groups require separatemanagement strategies, and have different responsesto treatment.
A pathological-pathophysiological classification ofcongenital systemic-to-pulmonary shunts associatedwith PAH has also been proposed to better describethe type of congenital shunts (Table 2).

NATURAL HISTORY-EISENMENGERSYNDROME
Surgical Correction/Operability
Advances in pediatric cardiac surgery now enablecorrective surgery for CHD that are associatedwith increased pulmonary blood flow to take placein very early infancy. These procedures aim toprevent a whole plethora of sequelae, including thedevelopment of PAH and PVD. However, in someindividuals with left to right shunts, such defectsmay pass undetected until childhood or evenadulthood and are diagnosed late when pulmonaryvascular lesions have developed. In developingcountries, due to a previous lack of opportunity toclose defects during infancy, PAH in children withCHD is common. This situation is now becominga very real issue, as healthcare in these countries isstarting to improve.11 Therefore, there is a real needfor guidance concerning complete surgical repairor palliative surgery of CHD in patients that, asa result of their condition, develop some degree ofPVD.
How can patients with a high risk of persistentPAH after surgery be identified with accuracy?Physicians currently use to base their decision ondifferent criteria on whether a patient is a suitablesurgical candidate. There is no overall consensus andrecommendations rather than definitive guidancecan only be given.12
Surgical repair in patients with high PVR andestablished PAH is risky. If PVR remains highpost-operatively and PAH persists then prognosis is poor.13 In a 5-year retrospective study ofchildren with PAH in the United Kingdom, thesubpopulation with post-operative CHD-PAHfared far worse than those with PAH associatedwith complex (un-operated) CHD and ES. Almostone quarter of these children died (11/47).13 Children with ES had a greater cumulative survivaltime by 1.3 years indicating that surgical repair isnot necessarily always the best option.
There are several examinations and criteriathat are used to inform the decision on whether apatient with PAH related to CHD is suitable forsurgery and ascertain the best possible outcomesthat can be attained: clinical examination for signsof congestive heart failure and oxygen saturation;echocardiography for signs of pulmonary over-circulation (Figure 1 and 2); and the current goldstandard of right heart catheterisation measurementsof hemodynamic parameters and vasoreactivity(Figure 3).11,14

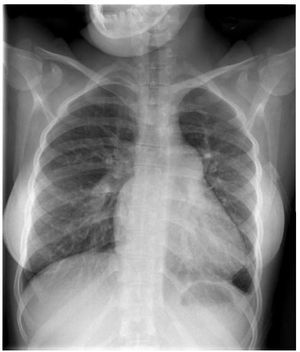
Figure 1. Chest radiograph of a patient with Eisenmenger syndromeshowing mild cardiomegaly, enlarged pulmonary arteries and decreasedperipheral pulmonary vascular markings. These signs prelude the possibilityof surgical repair.

Figure 2. Echocardiography of a patient with a ventricular septal defects and signs of pulmonary overcirculation attesting for low pulmonary vascularresistance and high pulmonary blood flow. These pictures are usually allows for surgical repair with performing catheterization. Left pannel shows a shortaxis view with the ventricular septal defect defect (arrow) and in particular a dilated left atrium (LA). Right panel shows a long axis view with a dilated leftventricle (LV) and LA attesting pulmonary overcirculation in presence of a shunt.

Figure 3. Ideal cardiac catheterizationwith catheters measuring simultaneouslys oxygen saturation and pressures in allcavities and vessels simultaneously toavoid time sampling bias. It describe theformula to calculate Qp/Qs in congenitalshunts which is the ration of pulmonaryblood flow (Qp) over systemic blood flow(Qs), Cardiac output can be measured bythermodilution in absence of shunt or withthe Fick formula in presence of intra orextracardiac shunts. LAP indicates meanleft atrial pressure; PAP, mean pulmonaryarterial pressure; PCWP, pulmonarycapillary wedge pressure; RAP, meanright atrial pressure. (Courtesy of IngramSchulze Neick National & UK Centre forPulmonary Hypertension in Children, GreatOrmond Street Hospital, London, UnitedKingdom).
For some time examination of histopathologicalchanges of the pulmonary vasculature by meansof lung biopsy was used to assess operability.15 At present, the reliability of the results is notconsidered sufficient to justify the invasive natureand risks involved in obtaining a tissue sample.16 Moreover biopsy may not represent the all lungdisease but just a random part. Patients withoutintimal thickening of the pulmonary arteriesand so considered to have reversible PVD canstill develop irreversible postoperative PAH.Moreover, younger children (<2 years) are often operable despite advanced changes on lungbiopsy.15 However, in light of new informationcorrelating apoptotic markers to morphologicalchanges in lung tissue and the development ofirreversible post-operative PAH,9 lung biopsies doand should continue to play an important role inclinical and basic research. A better understanding of the pathophysiology of the pulmonaryvasculature in PAH-CHD is still required in orderto fully examine the effects drugs and treatmentsavailable now and in the future. See Viswanathanand Kumar,11 Lopes and O'Leary,12 and Giglia andHumpl14 for comprehensive reviews on invasiveand non-invasive assessments for determiningoperability in PAH-CHD patients.
At present, empirical means using hemodynamicdata from right heart catheterisation andvasoreactivity are principally used to best predictwhich patients would have a positive or negativesurgical outcome. In a recent paper, Lopesand O'Leary, from available literature and byseeking expert opinion from recognised centresof excellence, specified hemodynamic criteriabased on both PVR and the ratio of pulmonaryto systemic resistance and the way these valueschange during acute vasodilator challenge. It wasdetermined that12:
1. A baseline PVR index <6 Woods units/m2 associated with a resistance ratio of <0.3 withouta vasoreactivity test is interpreted as indicative of afavourable outcome following operations resultingin a biventricular circulation.
2. Acute vasodilator challenge using oxygen/nitric oxide has been strongly encouraged if baselinePVR index is between 6 and 9 Wood units/m2 in thepresence of a resistance ratio from around 0.3-0.5.Although there is no absolute consensus, operabilitywith a favourable outcome is considered likely if thefollowing criteria are met:
- A decrease of 20% in the PVR index.
- A decrease of around 20% in the ratio ofpulmonary to systemic vascular resistance.
- A final PVR index of <6 Woods units/m2.
- A final ratio of resistance of <0.3.
These are very conservative numbers thatmay be adapted in the future. Acute vasodilatortesting, regardless of whether nitric oxidealone or in an admixture with oxygen, is thegold standard measure of the reactivity of thepulmonary vascular bed.17,18 In idiopathic PAH, vasodilator testing is done in order to determinewhether a patient will respond to therapy withcalcium channel antagonists. However, when toassess the operability of a patient with a CHDand high PVR, it is an open question on whetherit is accurate enough to completely discriminatebetween patients who will or won't have a goodsurgical outcome. In addition, technical difficultiesleading to calculation errors and other medicalconditions need to be considered when undertakingvasodilator testing.11 It remains unclear whichpreoperative pulmonary hemodynamic parametercorrelate with the best outcomes. How individualpatient factors such as cardiac lesion type andgenetic predisposition influence the outcome isalso not completely understood.
It should be noted that the above criteria do notapply to patients with single ventricle physiologywho are being assessed for the creation of a Fontancirculation. These patients should ideally havenear normal levels of PVR and certainly no morethan 3 Woods unit per m2. Moreover, obtaining accurate hemodynamic measurements can be evenmore difficult in patients with single ventriclephysiology.14
Although right heart catheterisationmeasurement of hemodynamics are helpful, andindeed the best tools available at present, they arenot failsafe. Patients that fall within the rangesdeemed appropriate for determining a goodoperable outcome may still present with persistentpost-operative PAH. Better, more accurate andpreferably less invasive evaluation tools are needed,and especially in patients considered borderlinefor operability due to their hemodynamicprofile. Recently, Lévy et al have made somepromising advances to develop new tools to assessoperability.19-21
Patients with similar hemodynamics priorto operation were stratified into two groupspost-operatively depending on the presence ofpersistent postoperative hypertension. In lungbiopsies, although increases in pulmonary arterialwall thickness were apparent for all patients, 10/11of those with irreversible pulmonary hypertensionalso had pronounced thickening of the intimallayer. Coupled with this was the exclusiveexpression of Bcl-2, an antiapoptotic marker,by the endothelial cells from arteries with severeintimal fibrosis. There was no difference betweenthe expression of apoptotic markers, capsase-3and p53, in the endothelial cells of the groups.These data suggest that proliferation of apoptotic-resistant endothelial cells may be a causative factorof intimal thickening. Experimental evidence fromanimal models of PAH supports a hypothesis thata triggering event leading to initial apoptosisof endothelial cells could promote subsequentproliferation and emergence of apoptotic resistantendothelial cells.
Recognising that lung biopsy is an invasive tooland less than ideal in general clinical practice,Smadja et al set out to determine whethercirculating endothelial cells (CEC)—alreadyrecognised as a non-invasive marker of vasculardamage, remodelling and dysfunction—would bea suitable biomarker that could identify patientsat high risk of developing irreversible PAH afterrepair of a CHD. Patients with irreversible PAH,in addition to showing pulmonary arterial intimalthickening and a corresponding high expression ofblc-2 in the endothelial cells on lung biopsy, alsohad significantly higher peripheral blood CEClevels than those with reversible PAH.20 In contrastto CEC, other biomarkers of endothelial activation,regeneration and injury have not been able todiscriminate between reversible and irreversiblePAH following surgery.
Non-operable preEisenmenger
As mentioned above the second group of thesubclassification includes patient with PVRconsidered too high for surgical repair but they donot reach the diagnosis of ES. So far, the therapeuticapproach of this group is to watch and see andwait for the development of ES. However, withthe upcoming new targeted therapies use to treatpulmonary hypertension in several settings, theconcept of treat and repair as emerged, with morequestions than answers!22
In the past 10 years prostacyclin analogues,endothelin receptor antagonists andphosphodiesterase type-5 (PDE-5) inhibitors haveall shown to be effective in treating PAH, in part viavasodilatory actions.23 It is proposed that endothelinreceptor antagonists in particular may have additionalactions such preventing endothelial cell growthand fibrosis and have a remodelling effect on thepulmonary vascular bed. Indeed, their vasodilatoryactions are of lesser interest in the discussion of thepotential ability to prepare PAH-CHD patients foroperability; it is their antiproliferative actions andpotential to induce lesion regression that is of greaterimportance.
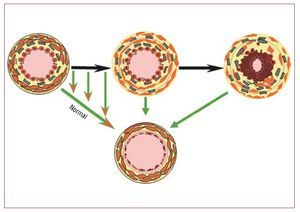
The dual endothelin receptor antagonistbosentan, has been extensively tested in idiopathicPAH and there is also good evidence that it iseffective in treating PAH associated with CHD. 24 Unlike idiopathic PAH, the cause of PAH in CHD patients with large defects is partially known; itis thought that the pressure and volume load onthe pulmonary vascular bed leads to pulmonaryvascular remodelling and lesions. While inidiopathic PAH patients always have definedlesions, this may not always be the case for CHDpatients where lesions may be less extensive.So by reducing PVR in patients where vascularlesions are not extensive, the possibility arises thatpre-treatment with vasodilators can be used toimprove a patient's condition and an inoperablecase could be considered operable (Figure 4). Thismay not, however, always be the case in patientswhere lesions are extensive and PVD is established.Although drugs can reduce PVR in these patients,PAH could remain post operatively and a worseprognosis could result.
Figure 4. Treat and repair? The upperpart shows the progression of lesionsin presence of a left to right shunt. Thearrows represent the opportunity to seethe lesions to regress to normal or nearnormal. This raises the opportunity totreat patients with increased pulmonaryvascular resistance that contraindicatessurgery in order to remodel the vascularbed and possible to allow completecorrection of the underlying anatomicallesion.
One problem that may arise by decreasing PVRwith PAH-targeted therapies is that an increase inpulmonary blood flow, due to an increase in shuntpressure, re-establishes the propensity towardslesion occurrence. Thus, paradoxically, the reversalof vascular remodelling and lesion formation,leading to an initial decrease in PVR could actuallyresult in pulmonary vascular damage later on. Onesolution would be to apply a pulmonary artery bandonce PVR decreases, thereby reducing blood flow tothe pulmonary vascular bed and preventing furtherdamage.
In addition to vasodilatory actions, bosentanalso has anti-fibrotic, anti-proliferative and anti-inflammatory actions. Prostacyclin analoguesinhibit platelet aggregation and smooth musclecell growth. The additional properties of thesedrugs may also have a role in preventing or slowingvascular remodelling. There have been severalcase reports of pre-treatment with prostacyclinsor bosentan being used prior to CHD surgery toprepare borderline or "inoperable patients."25-28 These reports suggest an advantage to usingprostacyclin analogues or endothelin receptorantagonists to improve hemodynamic and makeconditions more favourable for repair. However,there are several very important elements that needto be considered. Most patients had simple atrialseptal defects (ASDs). Assessment of operabilitymay be questionable. For most of these case reportsthe follow-up time of the patients when outcomeswere reported was short—1 year or less—. Toconfidently declare a successful outcome, data overa minimum of several years would be required.While these isolated cases show success with pre-treatment prior to operability, we do not knowhow many cases have failed and not been reported.A retrospective analysis of national registries onthis type of data would be required in order to gaina complete picture.
PERSISTENT PULMONARY HYPERTENSIONLATE AFTER SURGICAL REPAIR
The functional and structural state of thepulmonary vascular bed plays a pivotal role inthe presentation and outcome of the child withcongenital cardiovascular disease. It is in theimmediate postoperative period that the child ismost vulnerable to a sudden or sustained increasein pulmonary vascular resistance. Following surgeryfor congenital cardiac disease, pulmonary vascularreactivity is heightened, and vasospastic stimuli mayresult in sudden increases in PAP and resistance,resulting in acute right-sided cardiac failure, tricuspidregurgitation, systemic hypotension, myocardialischemia, and increased resistance in the airways.29 These episodes, called pulmonary hypertensivecrises, may be lethal events. Mildly stimulatingevents can precipitate similar crises, and the crisestend to last longer and cluster.
However, with improved postoperative care andthe introduction of new therapies, acute pulmonaryhypertensive crisis can, in most cases, be treated.
The incidence of postoperative pulmonaryhypertensive events decreased from 31% in theperiod from 1980 through 1984, to 6.8% before theroutine use of inhaled nitric oxide.30 Series reflectiveof contemporary practice suggest that pulmonaryhypertension complicates 2% of patients undergoing congenital cardiac surgery, with crises occurringin 0.75%.7 The mortality in those suffering a crisis,nonetheless, remains high at 20%, and pulmonaryvascular disease is identified as a major contributorto length of stay in hospital, and the need forprolonged mechanical ventilation (see a recentreview by Adatia and Beghetti for further detail, asthis is not the scope of this review.29)
However, acute treatment and survival in theimmediate postoperative period does not mean thatpulmonary hypertension resolves and the patientmay present persistent PAH after surgical repair butin the absence of a shunt. Little is known about thisparticular group, as literature remains scarce. Thisgroup has been included in the subclassification ofCHD-PAH (group 4 of the clinical subclassification).6 The hemodynamic presented by this group is verysimilar to idiopathic PAH and thus prognosis seemsto be poor based on recent data.13 This underscoresthe importance of accurate decision for operabilityand survival could even be better with an open shuntand ES than with a closed shunt and right ventricularfailure.31
EISENMENGER SYNDROME MANAGEMENT
ES represents the most advanced form of PAHassociated with CHD. The signs and symptoms ofES usually result from low blood oxygen saturation and include dyspnoea, cyanosis, fatigue, dizziness,syncope, and arrhythmia. Symptoms may not ariseuntil childhood or early adulthood. In general,patients with ES have reduced life expectancy,although many survive into their third or fourthdecade,32 with some even surviving into theirseventh decade with appropriate management.32,33 Of all patients with CHD, those with ES are themost severely compromised in terms of exerciseintolerance.34 Exercise intolerance in these patientshas been identified as a predictor of hospitalisationor death independent of age, gender, functionalclass or underlying cardiac defect.34 Anecdotalevidence suggests that patients with ES adapt theirlifestyle around their exercise capabilities, and thatthey tend to under-report their limitations. Despitethis, ES clearly and severely affects a patient'sexercise capacity and so decreases their quality oflife.
Conventional Management
Recent publications have extensively describedthe management of ES.2,5,35 As mentioned earlier, treatment options for patients with ES werehistorically limited to palliative measures andheart-lung transplantation. Treatment mostcommonly involved the use of digitalis, diuretics,antiarrhythmics, and/or anticoagulants. However,none of these classes of drugs significantly modifiessurvival or significantly affects the risk of deteriorationin ES. Digoxin has been used in palliative therapyfor right heart failure in ES, although availableevidence supporting its use is particularly weak.36 The use of anticoagulation in patients with ES iscontroversial, as ES patients have a high incidence ofpulmonary artery thrombosis, haemoptysis, strokeand an increased risk of haemorrhage.36 One recent study estimated the prevalent pulmonary arterythrombosis in ES to be 20%, with risk correlating withincreasing age, biventricular dysfunction, dilatationof the pulmonary arteries, and concomitantlydecreased pulmonary flow velocity.37 Althoughevidence suggests a benefit of such treatment inpatients with idiopatic PAH, no data exist in ES,and the haemorrhage associated risks of treatmentin these patients may outweigh potential benefits.To date, no prospective studies have addressed thevalue of anticoagulation in the prevention of eitherthrombosis or haemoptysis, and a great need existsfor such data.38
The efficacy of calcium channel blockers inpatients with ES is neither proven nor generallyrecommended, as their use can result in an acutedecrease in systemic arterial pressure and increasedright-to-left shunting, which may lead to syncopeand sudden death.39 Long-term oxygen therapy at home for a minimum of 12-15 hours per day mayimprove symptoms, but has not been shown tomodify survival.36
Patient education, behavioural modifications andawareness of potential medical risk factors are allimportant aspects of management. Patients withES are at particular risk during cardiac and non-cardiac surgery and anaesthesia, and as a resultof dehydration, chest infections, high altitudeand intravenous lines. It is also recommended toavoid strenuous exercise and not to participate incompetitive sports.
Pregnancy is associated with high risk to bothmother and foetus. Spontaneous abortion ratesare high and only around 25% of pregnanciescontinue to term. For those infants who survive,around one third have evidence of intrauterinegrowth retardation, and perinatal mortality is high.Maternal mortality is around 45% in ES patients,and death usually occurs during delivery or withinthe first week postpartum, mostly as a result ofthromboembolism, hypovolaemia, or preeclampsia . Pregnancy is therefore contraindicated in patientswith ES.40,41
Targeted Therapies
As discussed, the endothelin-1 system clearlyplays a major role in the structural and functionalabnormalities in the pulmonary vasculatureand the progression of PAH in all forms of thecondition, including PAH-CHD. Given thattreatment with endothelin receptor antagonists hasbeen successful in treating patients with idiopathicPAH and PAH-CTD,42-44 they would be expected to have similarly beneficial effects in patients withPAH-CHD. The first randomised, double-blind,placebo-controlled study in patients with ES wasBREATHE-5 (Bosentan Randomised Trial ofEndothelin Antagonist-5) which investigated theefficacy of the dual endothelin receptor antagonist,bosentan, in 54 adult patients with ES. During this16-week study, bosentan significantly reduced PVRand mean PAP and improved exercise capacitycompared with the placebo group, without adverselyaffecting systemic arterial oxygen saturation.45 This safety finding is of particular importance inpatients with ES given the potential for aggravationof the overall shunt due to the possibility of a fallin systemic resistance in response to vasodilatorytherapies. Longer follow-up data from the open-label follow-up study of patients enrolled in theoriginal 16-week double-blind trial showed thatimprovements in exercise capacity continued overa further 24 weeks of treatment.46 Functional classalso improved over this period, and treatment waswell tolerated.
These findings are supported by a numberof small-scale, open-label studies, which alsodemonstrate improvements in functional class,oxygen saturation, clinical status and pulmonaryhemodynamic in paediatric and adult patientswith ES.47-50 Long-term data suggest thatimprovements are maintained for as long as twoyears of treatment, without safety or tolerabilityissues51,52 but other have shown that the effect may decrease with time.53 The findings from thesestudies challenge the dogma that pulmonaryvascular disease in patients with ES is notamenable to treatment. On the other hand, ESis not a stable disease as has been assumed, butprogressive deterioration occurs, as demonstratedby the increase in PVR seen in patients from theplacebo arm of the BREATHE-5 study.45
Data from the BREATHE-5 study alsosuggest that the location of the septal defectdoes not influence short-term hemodynamic andfunctional improvements to bosentan treatment.As the evolution of pulmonary vascular diseasediffers markedly between ES patients with ASDsand ventricular septal defects (VSDs), they couldrespond differently to medical treatment. In apost hoc analysis of the BREATHE-5 trial, theeffects of bosentan and placebo in patients withASD and patients with either VSD or both defects(VSD/ASD+VSD) were compared.54 In both subgroups, no changes in systemic pulse oximetrywere observed between treatment groups, andplacebo-corrected treatment effects on indexedPVR, exercise capacity and mean PAP were alsocomparable.
Data with other endothelin receptor antagonists inPAH-CHD are still awaited but it would be expectedto see similar results as with other forms of PAH.
Phosphodiesterase Type 5 Inhibitors
To date, there are limited data regarding theuse of the PDE-5 inhibitors in patients with ES.After six months of treatment, World HealthOrganization (WHO) functional class, oxygensaturation, mean and systolic PAP and PVR weresignificantly improved in seven patients with ESwho participated in a prospective, open-label trialof sildenafil, but although there was a trend towardsimprovement, changes in 6 minute walk distance didnot reach significance.55 There were few significantside effects, and, although there was a theoreticalpossibility of reduced pulmonary blood flow dueto reduced systemic vascular resistance, cyanosisactually improved in these patients. Functional class,exercise capacity and pulmonary hemodynamicalso improved without significant side effects in21 patients with ES treated with sildenafil in a prospective, non-randomised, uncontrolled, dose-response trial.56
These findings are supported by other smallstudies of PDE-5 inhibitors, alone and incombination with prostanoids, which also showedimprovements in exercise capacity, functionalclass and some hemodynamic parameters withoutsafety issues.57-60 Recently, Tay et al showed that 3months sildenafil was well tolerated in 12 adult ESand associated with a significant improvement inquality of life and exercise capacity.61 Results withregard to efficacy, although encouraging, needvalidation in large randomised, placebo-controlledtrials. Such a trial—to investigate the effects ofsildenafil on exercise capacity and cardiopulmonaryhemodynamic in patients with ES—is currentlyrecruiting participants in Germany. In addition,long-term effects and effectiveness in ES patientswith more complex underlying defects remain to beestablished.
Prostacyclin and Prostacyclin Analogues
Overall, there are few data and no large trialsconcerning the use of prostanoids in ES. Long-termintravenous prostacyclin improved hemodynamicand functional class in 20 patients with PAHassociated with a range of CHD, although noneof the patients had an acute hemodynamicresponse.62 Continuous intravenous epoprostenolsignificantly improved functional class, arterialsaturation and 6-minute walk distance, anddecreased PVR in eight patients with ES afterthree months of therapy.63 However, in an earlierseries, treatment of patients with epoprostenolresulted in adverse events including increase insystemic vascular resistance, increased PVR andlow arterial oxygen in eight out of 10 patients.64 In addition, a number of adverse events have beenrecorded, including cerebrovascular accidentswhich probably resulted from the use of a centralvenous catheter in the presence of a right-to-leftshunt.63 Given the relatively longer median survivalin patients with ES relative to those with idiopaticPAH, the potential risks of long-term catheteruse is especially important when analysing therisk-to-benefit ratio of treatment. Data on otherforms of prostanoid therapy including inhaledand intravenous iloprost, and oral beraprost inES are limited to case reports, case studies, andsmall series. Inhaled and oral prostanoids offerobvious advantages over epoprostenol in terms ofthe safety of long-term administration, but theirefficacy and safety has not yet been fully studiedin this patient population.
A recent report of Dimopoulos et al showsimproved survival in ES using different type of targeted therapies and confirms the potential of thisapproach in ES.65
Thoracic Organ Transplantation
Ultimately, transplantation, preferably heart-lungtransplantation (HLT), is an option only for a small,selected subgroup of patients, and is severely limitedby the availability of donor organs. The success oftransplantation varies depending on the underlyingcause of ES, and appears to be most beneficialfor patients with VSD or multiple congenitalabnormalities.66 Overall, transplantation in patientswith ES is associated with high perioperativemortality.67 However, studies suggest that, althoughthe postoperative course tends to be complicatedin these patients, short- and long-term survivalrates following HLT are similar to those reportedin non-ES recipients.68 One-year survival rates ofapproximately 70% after HLT, and 55% after lungtransplantation, have been achieved. Five- and 10-year survival rates are 51% and 28%, respectively,after HLT.66,68
Given the paucity of suitable donor organs,the small number of suitable recipients and thepoor prognosis following HLT, any means todelay the need for HLT in ES patents would bevery welcome. One recent retrospective analysissuggests that ES patients who received novel,advanced therapies including prostacyclinanalogues and endothelin receptor antagonistsmay benefit from significantly longer mean timesto death or inscription on the active waiting list,by comparison with patients without.69 Given thelack of benefit of conventional therapy in ES,and the limited surgical options once the diseasehas developed, a clear unmet medical need existsfor patients with ES, which may be addressed bytargeted therapies.
The Fontan Circulation
Since its first description over three decadesago, the Fontan operation and its variations havebecome the procedures of choice in the managementof patients with congenital heart disease with asingle anatomical or functional ventricle. The aimof Fontan surgery is to use this single ventricle todrive the systemic circulation while the pulmonarycirculation is primarily driven by the negativeintrathoracic pressure.
Currently there is no satisfactory general medicaltreatment for failing Fontan. Management hasinvolved treating specific manifestations such asventricular dysfunction, protein loosing enteropathyand increased PVR. Treatment of ventriculardysfunction in Fontan patients has been attempted using a variety of agents, but there are few supportivedata, and there is evidence that these generally havelittle or no benefits due to lack of impact on reducedpreload.70 Angiotensin-converting enzyme inhibitorshave no effect on exercise capacity, systemic vascularresistance, resting cardiac index, or diastolic functionin Fontan patients.71 Despite this lack of evidence,however, many long-term Fontan patients aretreated.
Low cardiac output, excessive hypoxemia, orprotein loosing enteropathy may all be clinicalmanifestations of increased PVR; therefore, itsprevention or management is potentially of criticalimportance. In the acute, post-operative phase,patients with increased PVR are treated with nitricoxide and supplemental oxygen. Inhaled nitric oxidereduces central venous pressure,72 mean PAP andtranspulmonary pressure gradient. Prostacyclinshave been rarely used in perioperative Fontanpatients, and there are few data available. Beraprostreduces mean PAP and PVR in pre-operative Fontancandidates with mild pulmonary hypertension,73 and epoprostenol has been shown to prevent therebound effect after inhaled nitric oxide cessationin the early post-operative phase.74 When oraladministration becomes possible, sildenafil, a PDE-5 inhibitor which acts as a vasodilator, is often usedin the post-operative period as it is perceived as safe,with fast onset and good effectiveness. However,sildenafil is not approved for this use and there areno published data to support its efficacy or safety inthis indication.
In the treatment of patients with failing Fontan,there have been few observations published todate describing the effects of PVR-lowering drugs.Treatment of late Fontan patients with inhaled nitricoxide reduces PVR, although it has no significanteffect on cardiac index.75 NO-dependent, cyclicguanosine monophosphate (cGMP)-mediatedpulmonary vasodilatation can also be enhancedusing sildenafil. A single dose of sildenafil hasbeen shown to improve exercise capacity andhemodynamic response to exercise in late, non-failing Fontan patients.76 There are also singlecase studies showing improvement in a patientwith plastic bronchitis and a patient with proteinloosing enteropathy folllowing sildenafil.77,78 The effect of sildenafil on exercise tolerance, ventricularfunction and quality of life is currently underinvestigation in children who have undergone theFontan procedure. There are currently no dataon the effectiveness of prostanoid therapy in thefailing Fontan.
The dual endothelin receptor antagonist bosentanhas been shown to improve exercise capacity,functional class, quality of life and hemodynamicparameters including PVR and PAP in patients with PAH. Long-term treatment with bosentanimproved symptoms and aortic oxygen saturation,WHO functional class, maximal and sub-maximalexercise capacity, Borg dyspnoea index, meanPAP, pulmonary blood flow and PVR in a patientwith plastic bronchitis following Fontan.79 In a recent small study, oxygen saturation improvedin 5/9 patients during a 16-week treatment periodwith bosentan.80 The single endothelin receptorantagonists ambrisentan and sitaxentan are also usedin the management of PAH; however, no data areavailable describing their use in Fontan patients.
Given the important role of the pulmonaryvascular circulation in Fontan physiology, thedemonstrated increase in PAP with age, and thecurrent lack of data there is a major requirement forclinical studies on the efficacy and safety of potentialtherapies for failing Fontan on which to base muchneeded management recommendations.
CONCLUSIONS
In conclusion, improvements in diagnosis andsurgical and medical management have changed the long-term survival prospects for patients withPAH-CHD, resulting in a significant increase inthe number of patients surviving to adulthood.Although there are some differences in aetiology,treatment response and survival, pediatric and adultpatients with PAH-CHD have essentially the samedisease; a complex condition with a natural historythat varies depending on the underlying cardiacdefect and multiorgan chronic adaptation to it.Short-term benefits of novel targeted therapies inPAH-CHD are increasingly clear, but long-terminvestigation in PAH-CHD populations is required.Of increasing interest is the management of patientswith complex congenital defects, and the timing andoperability of patients to optimize outcomes, each ofwhich warrants further investigation. In particularthe role of targeted therapies and the potential treatand repair concept requires further studies. Anotherchallenge is the Fontan physiology that does notfulfil the diagnosis of PAH but where the role of thepulmonary circulation is central. Even, if progresshas occurred, still a lot of work is required to betterunderstand this problem in order to better addressPAH in this population.
ABBREVIATIONS
CEC: circulating endothelial cell
CHD: congenital heart disease
ES: Eisenmenger's syndrome
HLT: heart and lung transplantation
PAH: pulmonary arterial hypertension
PAP: pulmonary arterial pressure
PVD: pulmonary vascular disease
PVR: pulmonary vascular resistance
Conflict of interest:
Professor Maurice Beghetti has served on advisory boards/consulting forPfizer, Actelion Pharmaceuticals, Bayer Schering, GlaxoSmithKline, INOtherapeutics, Eli Lilly, and Mondobiotech and has received lecture feesfrom Actelion Pharmaceuticals, Encysive, Pfizer and Bayer Schering.
Correspondence: Prof. Maurice Beghetti, MD,
Pediatric Cardiology Unit. Department of the Child and Adolescent.Children's Hospital. University of Geneva,
6 rue Willy-Donzé. CH-1211 Geneva 14. Switzerland
E-mail: Maurice.Beghetti@hcuge.ch